



Inherited macular dystrophy represents a group of genetic disorders resulting in progressive damage to the cells in the macula leading to vision loss.
Inherited macular dystrophies are caused by mutations in genes that are essential for the function and survival of the cells in the macula. These mutations can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.
Overview of inherited macular dystrophies
Examples of autosomal dominant inherited macular dystrophies include Best disease and Stargardt disease; autosomal recessive inherited macular dystrophies include retinitis pigmentosa and Leber congenital amaurosis; X-linked inherited macular dystrophies include X-linked retinoschisis and X-linked cone dystrophy.
The symptoms of inherited macular dystrophies usually appear in childhood or adolescence but can also develop later in life. The initial symptoms may include difficulty seeing in low light, blurred or distorted vision, and decreased color vision. Central vision loss can occur as the condition progresses, leading to legal blindness.
There is currently no cure for inherited macular dystrophy, but treatments are available to slow down the progression of the disease and manage symptoms. These treatments may include nutritional supplements, low-vision aids, and gene therapy. Early diagnosis and management of symptoms are crucial to slow the disease's progression and preserving vision.
Common inherited macular dystrophies
Stargardt disease
Stargardt disease, also known as Stargardt macular degeneration or juvenile macular degeneration, is a rare genetic disorder caused by mutations in the ABCA4 gene, which encodes a protein that helps to remove toxic waste products from the retina.1
Clinical features
Stargardt disease typically presents in childhood or adolescence, although it can sometimes present in adulthood. The initial symptoms of Stargardt disease include blurry vision, difficulty reading, and sensitivity to bright light. As the disease progresses, central vision loss can occur. Other symptoms may include scotomas in the central vision, metamorphopsia, and difficulty recognizing faces.
Testing for Stargardt disease
Stargardt disease can be diagnosed through a comprehensive eye exam, which may include visual acuity testing, dilated fundus examination, electroretinography (ERG), optical coherence tomography (OCT), and fundus autofluorescence imaging (FAF).
In Figure 1A, the fundus photograph shows paramacular yellowish flecks and “beaten-bronze” central macular atrophy. In Figure 1B, fluorescein angiography of the same eye shows a dark choroid, hyper fluorescence associated with the flecks, and a bull’s-eye pattern of macular transmission defect. In images 1C and 1D, fundus photographs of the right and left eye show classic multiple, discrete, yellowish, pisciform (fish-shaped) flecks scattered throughout the posterior pole.

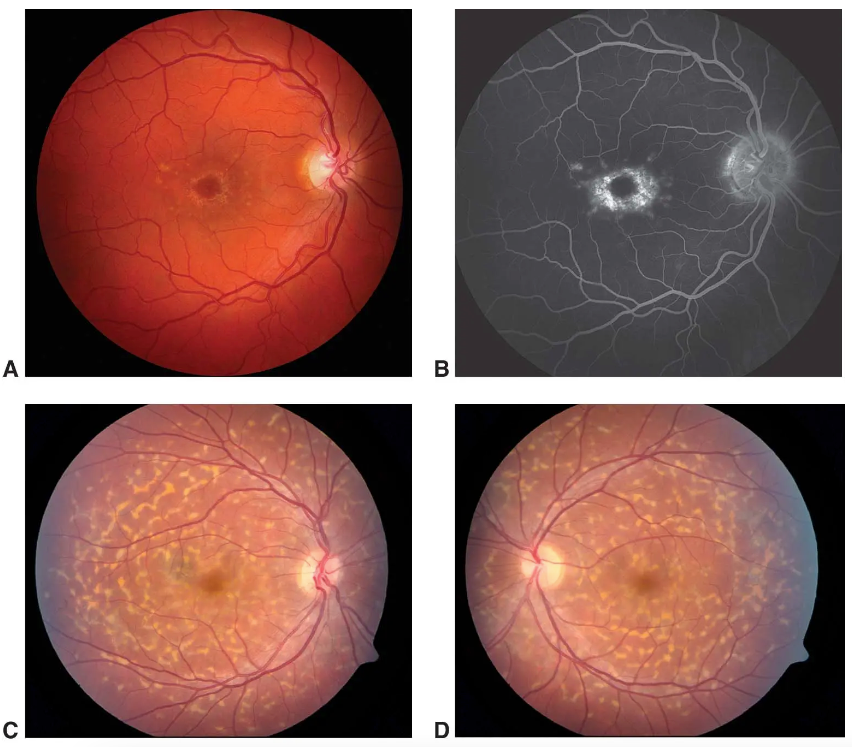
Figure 1: Courtesy of Mark Johson MD and Paul Sternberg MD.
Genetic inheritance of Stargardt disease
Stargardt disease is inherited in an autosomal recessive manner, meaning a person must inherit two copies of the mutated ABCA4 gene, one from each parent, to develop the disease. Genetic testing can be performed to identify mutations in the ABCA4 gene in affected individuals and their family members.1
Management and treatment of Stargardt disease
There is currently no cure for Stargardt disease. Still, several management and treatment options are available to slow down the progression of the disease and manage the symptoms.
These may include nutritional supplements, such as vitamins A, C, and E, which can help to slow down the accumulation of toxic waste products in the retina. Low vision aids, such as magnifying glasses, may also help manage the visual impairment associated with Stargardt disease. Experimental treatments, such as stem cell and gene therapy, are also being investigated as potential treatments for Stargardt disease.
Long-term prognosis of Stargardt disease
The long-term prognosis of Stargardt disease varies from person to person and depends on the severity and progression of the disease. In general, however, Stargardt disease tends to progress slowly over several years, eventually leading to legal blindness in most cases.
However, some individuals may retain good visual acuity well into adulthood, while others may experience rapid disease progression and vision loss in early childhood. Regular monitoring of the disease by an ophthalmologist is important for the early detection of any changes and adjustment of management and treatment options as necessary.
Download the Inherited Macular Dystrophies Cheat Sheet

Inherited Macular Dystrophies Cheat Sheet
This cheat sheet features annotated clinical images and foundational information on how to identify and treat inherited macular dystrophies.

Best disease
Best disease, also known as vitelliform macular dystrophy or Best vitelliform macular dystrophy (BVMD), is caused by mutations in the BEST1 gene, which encodes a protein in transporting ions and fluids across the retinal pigment epithelium (RPE) cells.2
Clinical features of Best disease
Best disease typically presents in childhood or adolescence. However, it can sometimes present in adulthood. Similar to other macular dystrophies, the initial symptoms of Best disease include decreased central vision, distortion, and difficulty with reading and recognizing faces.
A hallmark of Best disease is the presence of a yellowish, egg-shaped lesion in the macula called a vitelliform lesion, which is visible on ophthalmic examination. The vitelliform lesion can progress over time, eventually leading to atrophic or fibrotic changes in the macula and significant vision loss.3
Testing for Best disease
Best disease can be diagnosed through a comprehensive eye exam, which may include visual acuity testing, dilated fundus examination, ERG, OCT, and FAF. Genetic testing can also identify mutations in the BEST1 gene in affected individuals and their family members.
Figure 2 demonstrates an egg yolk lesion with satellite lesions caused by Best disease.

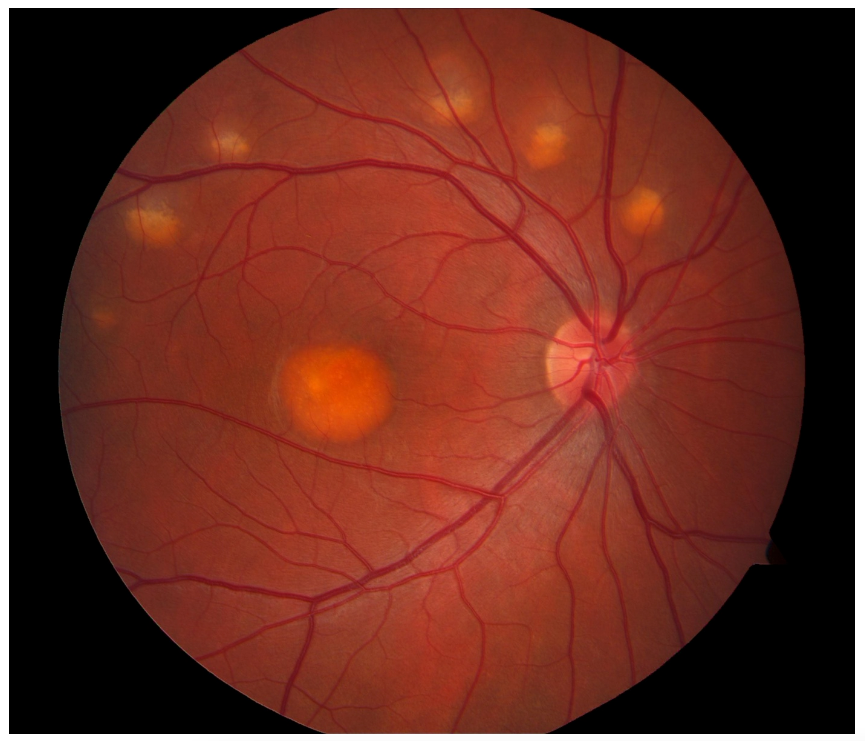
Figure 2: Courtesy of American Academy of Ophthalmology.
Genetic inheritance of Best disease
Best disease is inherited in an autosomal dominant manner. A person only needs to inherit one copy of the mutated BEST1 gene from one parent to develop the disease. Genetic testing can be performed to identify mutations in the BEST1 gene in affected individuals and their family members.3
Management and treatment of Best disease
There is currently no cure for Best disease. Like Stargardt, several management and treatment options are available to slow down the progression of the disease and manage the symptoms. Experimental treatments, such as gene and stem cell therapy, are also being investigated as potential treatments.
Long-term prognosis of Best disease
The long-term prognosis of Best disease varies from person to person and depends on the severity and progression of the disease. However, Best disease tends to progress slowly over several years, eventually leading to legal blindness in some cases.
Some individuals may retain good visual acuity well into adulthood, while others may experience rapid disease progression and vision loss in early childhood. An ophthalmologist's regular monitoring of the disease is important for the early detection of any changes and adjustment of management and treatment options as necessary.
X-linked retinoschisis
X-linked retinoschisis (XLRS) is a rare genetic disorder affecting the retina, the eye part responsible for vision. XLRS primarily affects males and is caused by mutations in the RS1 gene on the X chromosome, which codes for the protein retinoschisin.4
Clinical features of XLRS
XLRS typically presents in childhood, although the severity of symptoms can vary greatly. Common symptoms include decreased central vision, difficulty with color perception, and metamorphopsia. XLRS may also cause a splitting or separation of the layers of the retina, called retinoschisis, which can be detected on ophthalmic examination.
Testing for X-linked retinoschisis
XLRS can be diagnosed through a comprehensive eye exam, including visual acuity testing, dilated fundus examination, optical coherence tomography, and electroretinography (ERG). Genetic testing can identify mutations in the RS1 gene in affected individuals and their family members.
Figure 3 highlights OCT imaging of a child with X-linked retinoschisis.

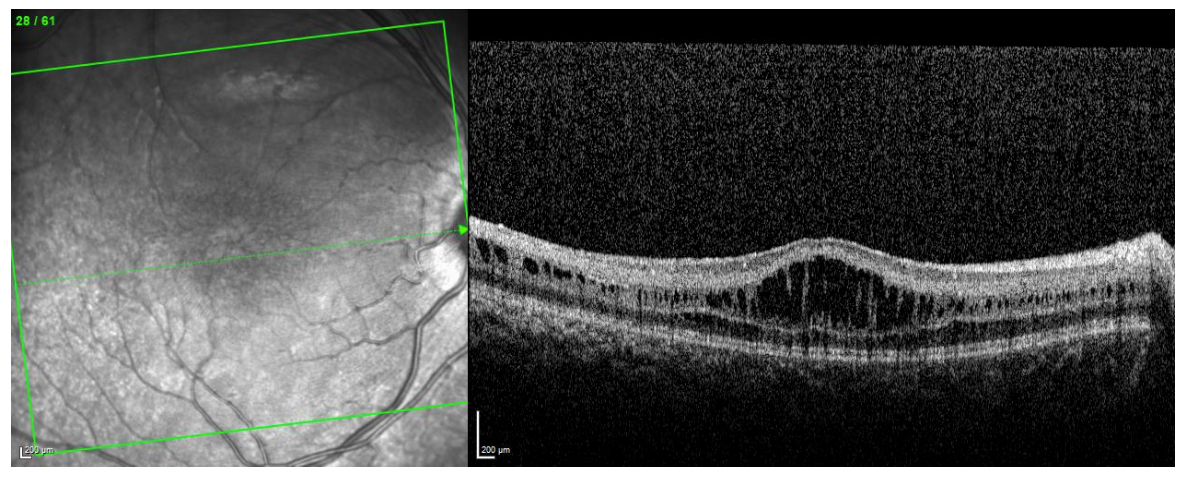
Figure 3: Courtesy of Andrew R. Lee, MD.
Genetic inheritance of XLRS
XLRS is inherited in an X-linked recessive pattern, meaning the RS1 gene mutation is on the X chromosome. As a result, males are more commonly affected than females. Female carriers of the RS1 gene mutation are typically asymptomatic but have a 50% chance of passing the mutation on to their offspring.4
Management and treatment of X-linked retinoschisis
Currently, there is no cure for XLRS. Still, there are several management and treatment options available to slow down the progression of the disease and manage the symptoms. Laser treatment or surgery may be used to treat retinoschisis.
However, these treatments are typically reserved for cases where retinoschisis causes severe vision loss secondary to retinal detachment or vitreous hemorrhage. Younger patients must be monitored closely for amblyopia. Topical or oral carbonic anhydrase inhibitors have shown promise in reducing schisis cavities and improving visual acuity.5
Long-term prognosis of XLRS
Like Stargardt and Best disease, the long-term prognosis of XLRS varies from person to person and depends on the severity and progression of the disease. As such, monitoring is important.
Sorsby macular dystrophy (SMD)
Clinical features of SMD
Sorsby macular dystrophy typically presents in adulthood and progresses rapidly, often leading to legal blindness within a few years. Common symptoms include blurred or distorted vision, difficulty seeing in low light conditions, and loss of central vision. SMD can also cause neovascularization leading to bleeding and scarring.6
Testing for Sorsby macular dystrophy
SMD can be diagnosed through a comprehensive eye exam, including visual acuity testing, dilated fundus examination, optical coherence tomography, and fluorescein angiography. Genetic testing can identify mutations in the TIMP3 gene associated with SMD.
Figure 4 shows the fundus photography of a 54-year-old woman with Sorsby macular dystrophy. Characteristic pale drusen are seen just outside the macula in both eyes. In 4A, there is fovea-sparing geographic atrophy in the left eye, and in 4B, the more severely affected right eye has subretinal fibrosis as well as geographic atrophy.

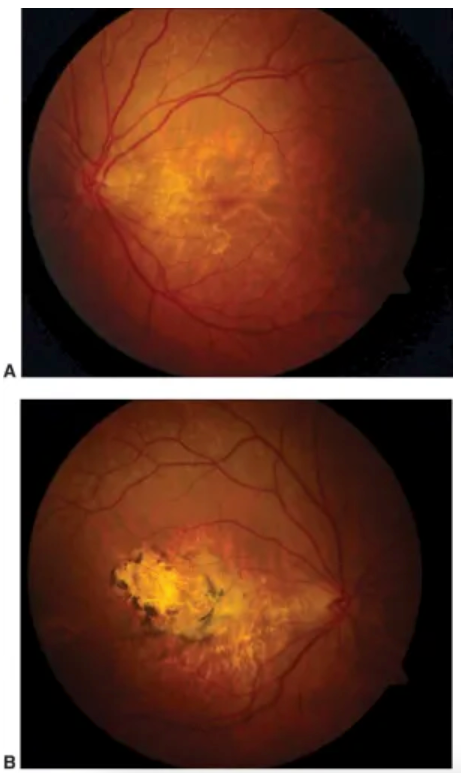
Figure 4: Courtesy of Cagri G. Besirli, MD.
Genetic inheritance of SMD
SMD is inherited in an autosomal dominant pattern, meaning that only one copy of the mutated TIMP3 gene is necessary to develop the disorder. Individuals with a family history of SMD have a 50% chance of inheriting the mutation from an affected parent.6
Management and treatment of Sorsby macular dystrophy
Currently, there is no cure for SMD, but there are several management and treatment options available to slow down the progression of the disease and manage the symptoms. Anti-VEGF injections and laser treatment can be used to treat neovascularization, which may help to preserve vision.6
Long-term prognosis of SMD
The long-term prognosis of SMD is generally poor, with most individuals experiencing rapid disease progression and legal blindness within a few years of diagnosis. However, the rate of disease progression can vary greatly from person to person, and some individuals may retain good visual acuity for several years.
An ophthalmologist's regular monitoring of the disease is important for the early detection of any changes and adjustment of management and treatment options as necessary.
Autosomal dominant radial drusen (ADD)
Clinical features of ADD
ADD typically presents in late childhood or early adulthood and is characterized by drusen in the macula.
Testing for autosomal dominant radial drusen
ADD can be diagnosed through a comprehensive eye exam, including visual acuity testing, dilated fundus examination, OCT, and fluorescein angiography. Genetic testing can confirm the presence of mutations in the EFEMP1 or TIMP3 genes, which are associated with ADD.7
In Figure 5, we see the honeycomb appearance of autosomal dominant radial drusen in the fundus.

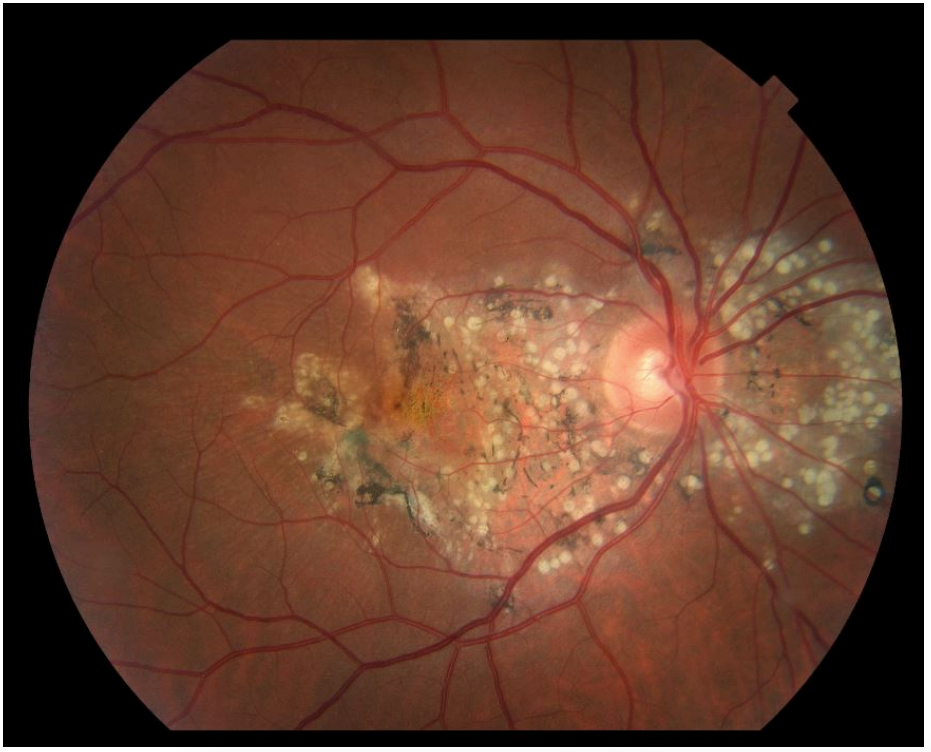
Figure 5: Courtesy of American Academy of Ophthalmology.
Genetic inheritance of ADD
ADD is inherited in an autosomal dominant pattern, meaning that only one copy of the mutated gene is necessary to develop the disorder. Mutations in the EFEMP1 or TIMP3 genes are associated with ADD, and individuals with a family history of the disorder have a 50% chance of inheriting the mutation from an affected parent.7
Management and treatment of ADD
Currently, there is no cure for ADD. Still, there are several management and treatment options available to slow down the progression of the disease and manage the symptoms, such as anti-VEGF therapy in the case of complication by choroidal neovascularization.
Long-term prognosis of autosomal dominant radial drusen
The long-term prognosis of ADD is generally good, with most individuals maintaining relatively stable visual acuity and quality of life. However, the rate of disease progression can vary greatly from person to person, and some individuals may experience a more rapid decline in vision than others.
An ophthalmologist's regular monitoring of the disease is important for the early detection of any changes and adjustment of management and treatment options as necessary.
Retinal pattern dystrophy
Retinal pattern dystrophies are a slowly progressive heterogeneous group of primarily autosomal dominantly inherited macular diseases that involves pigment deposition in the retinal pigment epithelium (RPE) of the macula.
Major categories or types of retinal pattern dystrophy include:
- Reticular dystrophy
- Fundus pulverulentus
- Butterfly-shaped pigment dystrophy
- Adult-onset foveomacular vitelliform dystrophy
- Multifocal pattern dystrophy simulating Stargardt disease.
Within the same patient, a given pattern may transform into another pattern, and each eye may have distinct patterns. Additionally, the same familial mutation can correspond to different phenotypic patterns.8
Clinical features of retina pattern dystrophies
Retinal pattern dystrophies are usually characterized by progressive vision loss, night blindness, and decreased color vision. The age of onset and the disease progression rate can vary widely among individuals and may be influenced by the specific genetic mutation involved. In some cases, retinal pattern dystrophies may also be associated with other ocular or systemic abnormalities.
Testing for retinal pattern dystrophies
Imaging studies, such as fundus photography, OCT, and fundus autofluorescence, are important for diagnosing retinal pattern dystrophies. These studies can help identify the characteristic pigment changes in the retina that are a hallmark of these conditions. Genetic testing is also an important diagnosis component, as it can identify the specific genetic mutations responsible for the disease.
Figure 6 illustrates FAF with increased and decreased autofluorescence corresponds to lipofuscin changes in butterfly pattern dystrophy.

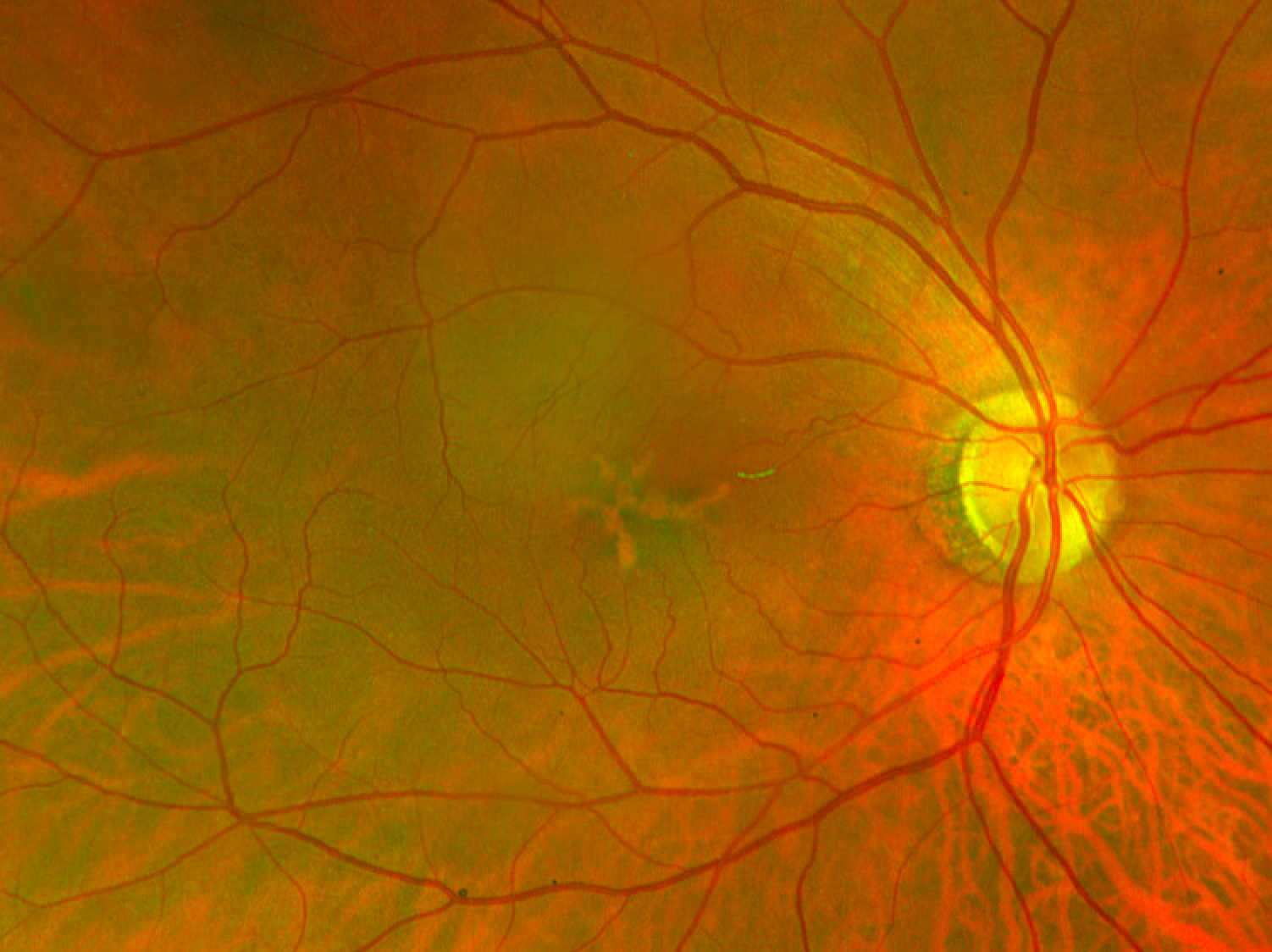
Figure 6: Courtesy of David Sierpina, MD.
Figure 7 demonstrates fluorescein angiography imaging of a multifocal pattern dystrophy.

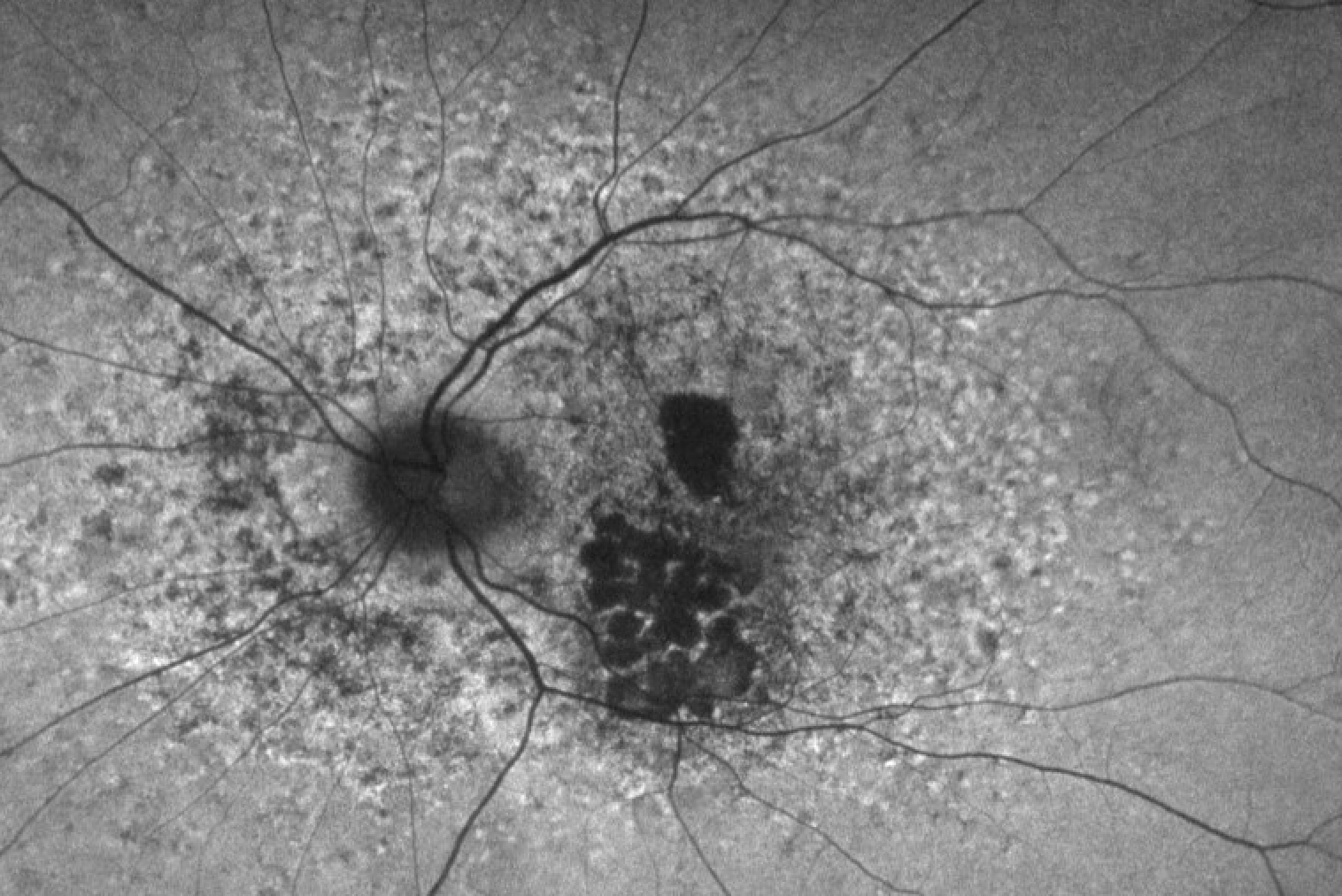
Figure 7: Courtesy of David Sierpina, MD.
Figure 8 features FAF demonstrating adult onset foveomacular vitelliform dystrophy.

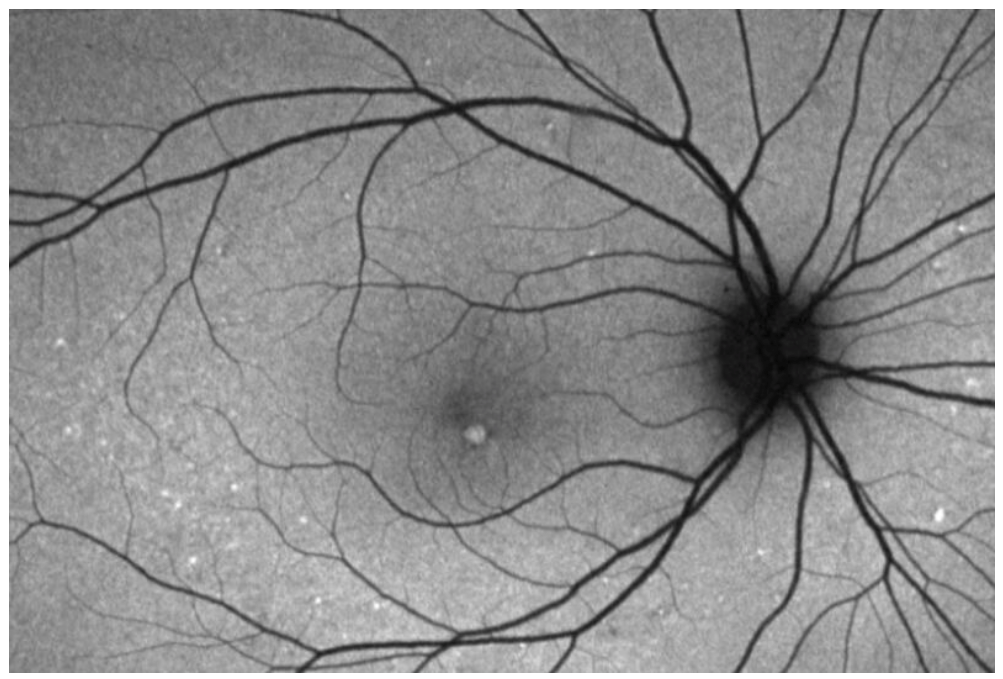
Figure 8: Courtesy of David Sierpina, MD.
Figure 9 demonstrates central areolar choroidal dystrophy in a patient with a heterozygous PRPH2 mutation (c.514C>T).
Figure 9 demonstrates central areolar choroidal dystrophy in a patient with a heterozygous PRPH2 mutation (c.514C>T).

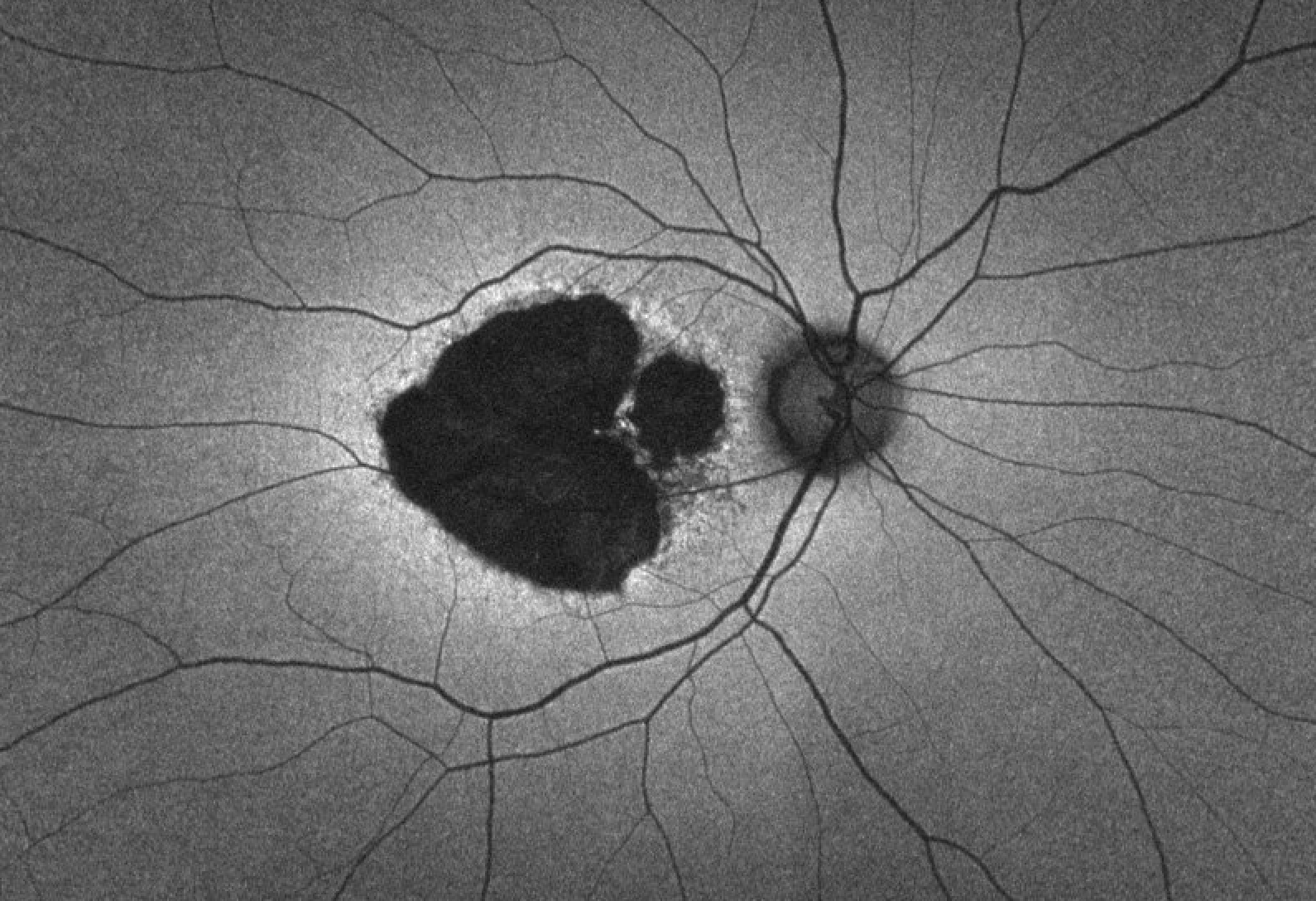
Figure 9: Courtesy of David Sierpina, MD.
Genetic inheritance of retinal pattern dystrophies
Retinal pattern dystrophies are inherited in an autosomal dominant or autosomal recessive manner, depending on the genetic mutation involved. Mutations in several genes have been identified as causes of retinal pattern dystrophies, including RDS, PRPH2, PROM1, and IMPG1. Sometimes, the specific genetic mutation may have implications for prognosis and treatment.8
Management and treatment of retinal pattern dystrophies
Currently, there is no cure for retinal pattern dystrophy, and treatment is focused on managing symptoms and preventing or slowing the progression of vision loss. This may include low-vision aids, such as magnifiers or electronic devices, to help improve visual function.
Genetic counseling is also an important management component, as it can help affected individuals and their families understand the inheritance pattern and make informed decisions about family planning. Secondary complications such as choroidal neovascularization can be treated with anti-VEGF injections.
Long-term prognosis of retinal pattern dystrophies
The long-term prognosis of retinal pattern dystrophies can vary widely depending on the specific genetic mutation involved and the age of onset. In some cases, vision loss may be gradual and relatively stable over many years, while in other cases, it may progress rapidly and lead to severe visual impairment or blindness.
Early diagnosis and management, including genetic counseling and low-vision aids, can help improve long-term outcomes and quality of life for affected individuals and their families.
The role of genetic testing in inherited macular dystrophies
These conditions are complex and can be difficult to diagnose, and genetic testing can provide critical information that can aid in accurate diagnosis and personalized treatment plans.
Genetic testing can help identify the specific genetic mutations causing a patient’s macular dystrophy. This information can provide insight into the underlying mechanisms of the disease and help guide treatment decisions. For example, certain genetic mutations may respond better to certain medications or therapies, and knowing a patient’s specific genetic mutation can help tailor their treatment plan accordingly.
In addition to aiding in diagnosis and treatment, genetic testing and databases can also play a critical role in the research and development of new treatments for macular dystrophies. Researchers can identify common genetic mutations and better understand the disease's underlying biology by compiling genetic data from many patients. This information can then be used to develop new treatments and therapies that target specific genetic mutations.
Using genetic testing to screen for inherited macular dystrophies
Furthermore, genetic testing and databases can also aid in the early detection and prevention of macular dystrophies. For individuals with a family history of macular dystrophy, genetic testing can identify whether they have inherited the genetic mutation that causes the disease. This information can be used to monitor their vision more closely and begin early interventions to prevent or slow disease progression.
It is important to note that genetic testing and databases must be used responsibly and with respect for patient privacy. Patients must give informed consent before undergoing genetic testing, and the testing results must be kept confidential and protected. However, with appropriate safeguards, the benefits of genetic testing and databases for diagnosing and treating macular dystrophies far outweigh the risks.
