




Inherited retinal diseases (IRD) represent a group of disorders that commonly result in progressive photoreceptor death leading to vision loss.1 IRDs display wide variation in their mode of inheritance, underlying genetic defects, age of onset, and functional severity.1 As the knowledge of genetics and the molecular mechanisms for IRD grows, the treatment options for these diseases expand.
In the past three decades, there has been an ever-increasing list of causative genes associated IRD.2 One of the first approved gene therapies available in the United States is voretigene neparvovec (Luxturna) used to treat RPE65-mediated Leber's congenital amaurosis (LCA).3
Genetic underpinnings of inherited retinal diseases
In the most basic definition, genes are composed of a promotor sequence, exons, and introns. The promotor controls how a gene is regulated, exons provide the genetic code for the resulting protein, and introns are intervening sequences that are spliced out as pre-mRNA.4 Mutations to these key elements produce alterations in the nucleotide sequence of the genome (Fig. 1).
The functional effect of these mutations is determined by the gene’s pattern of inheritance and can result in genes that have a loss of function, where the protein coded has reduced or no activity; gain of function, where the protein acquires harmful new function; or dominant-negative mutations, where mutated proteins interfere with its normal version.5

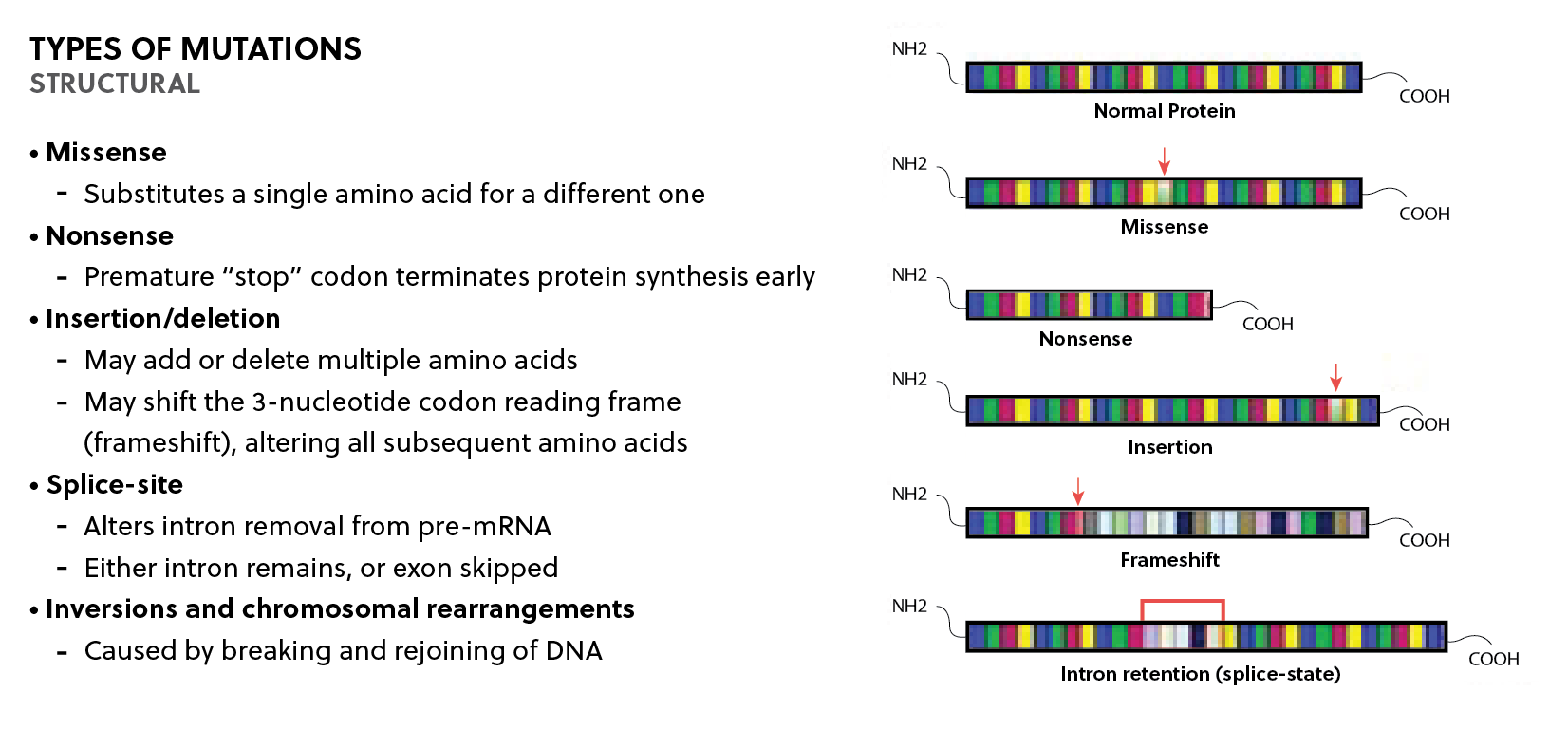
Figure 1: Summary of the different types of structural mutations relevant to inherited retinal diseases. Mutations in critical gene elements produce alterations in the nucleotide sequence of the genome with resultant deleterious effects on protein function.
In disease processes, a mutation in a single gene is capable of causing disease (monogenic); however, there are also numerous diseases where multiple gene variations cause morbidity (polygenic).5 In monogenic disorders, the pattern of inheritance may be de novo, or follow an autosomal dominant, autosomal recessive, X-linked dominant, X-linked recessive, or mitochondrial pattern.5 The physiological manifestation, or phenotype, of these inherited mutations may be variable due to expressivity, other gene alleles influencing phenotype, and environmental factors.5 Additionally, some genes have variable penetrance, where the probability of a disease manifesting when a disease genotype is present is not always certain.5
IRDs represent a large group of genetically and phenotypically heterogeneous retinal disorders. Collectively, they are the most prevalent inherited forms of visual handicap with an estimated prevalence of 1 in 300.6 Within IRD, there is a wide variation in the mode of inheritance; however, monogenic inheritance patterns exist, and can be autosomal dominant (e.g. types of retinitis pigmentosa), recessive (e.g. LCA type II), X-linked (e.g. retinoschisis) or a mitochondrial inheritance pattern (e.g. Leber's hereditary optic neuropathy).7 Many genes have now been identified to be associated with either isolated retinal disorders or systemic diseases involving the retina.2
Testing for these disorders commonly uses gene panels as well as single-gene and whole genome/exome testing (Fig. 2).

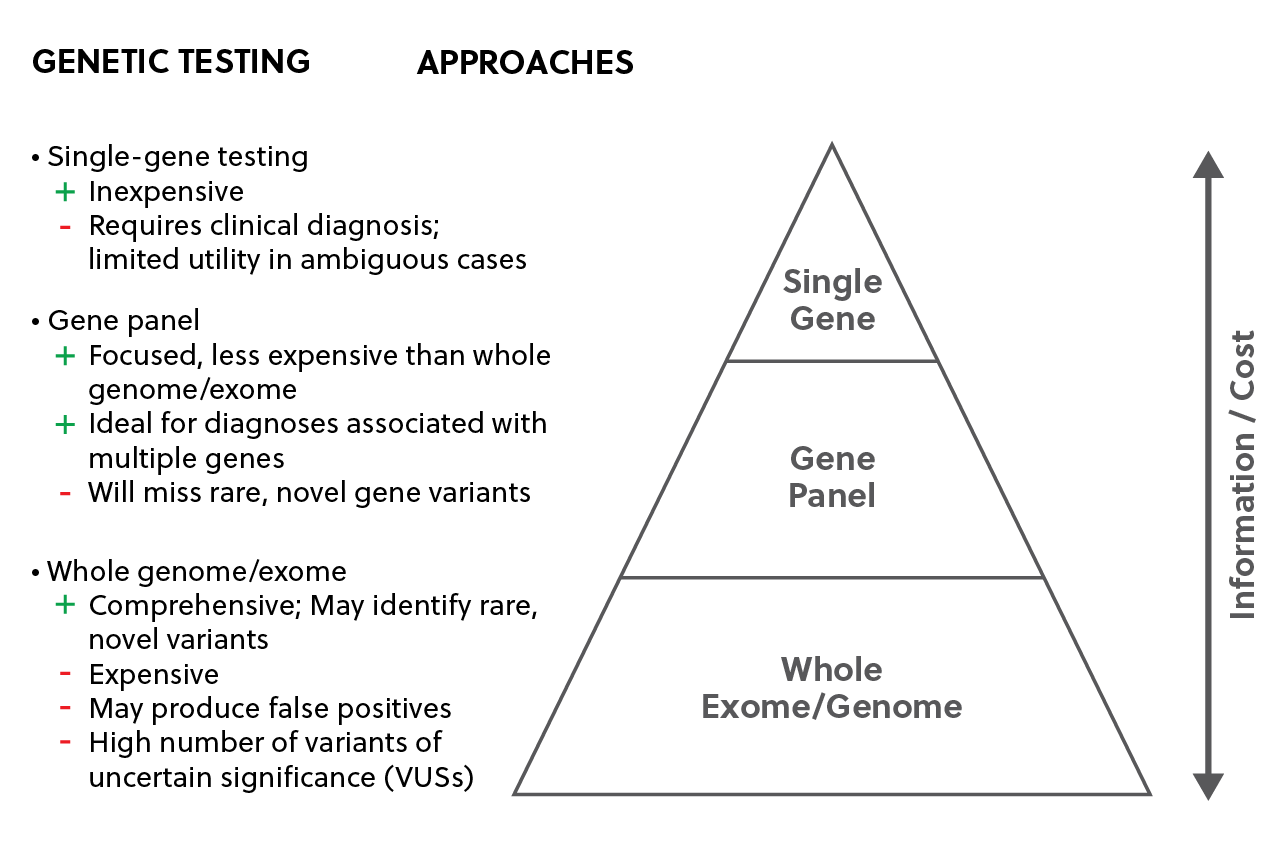
Figure 2: Genetic testing approaches for inherited retinal diseases including single-gene testing, gene panels, and whole genome/exome assays.
Gene therapy for IRD
Gene therapy is a therapeutic technique that involves the delivery of genetic material to modify gene expression or to correct an abnormal gene.4 Over 2600 gene therapy clinical trials are planned, ongoing, or have been completed worldwide, some of which are under development to treat monogenic IRD.8 There are several ways to introduce corrective genetic material into cells to compensate for a specific diseased gene. These include gene augmentation, gene inactivation, and gene editing.4
In addition to these techniques, facilitation with the use of specialized gene delivery vectors is often used to increase therapeutic efficiency and treatment duration.4 The most prevalent vector used in gene therapy is viral vectors, which are modified viruses with viral DNA removed.9 These vectors provide efficient cell entry and transit to the nucleus and can target specific cell types.9
In many cases of X-linked and recessive diseases such as Stargardt or X-linked retinoschisis, there is a decreased production of protein, absence of protein, or production of functionally null protein.10 Here, gene augmentation is the most commonly used straightforward technique. Gene augmentation strategies involve the insertion of an additional functional copy of an existing gene to restore or enhance wild-type protein expression.4 There are clinical trials evidencing the ability of gene augmentation to significantly ameliorate disease phenotype in both Stargardt, LCA, and X-linked retinoschisis.10
In the phase III trial outlining the use of Luxturna for LCA, results show clinically significant improvements in mean functional vision (measured by multi-luminance mobility test) with no immune-related adverse events.11 Further clinical trials may also highlight the importance of appropriate dosage and early intervention for implementing factors for better rescue and avoiding undesirable effects in bystander cells.
Approximately one-third of all IRDs are inherited in an autosomal dominant fashion, such as retinitis pigmentosa, where there is a gain-of-function mutant allele.10 Often, a combined approach is warranted, utilizing both gene inactivation and augmentation therapies.12 Clinical trials have shown evidence for strategies where initially a gene suppressor is delivered to down-regulate and inactivate the target, at which point a separate vector can then deliver the functional replacement gene.12
Additionally, gene editing has become an emerging and novel approach to disorders with large causative genes. The use of CRISPR (clustered regularly interspaced short palindromic repeats) to precisely cut and remove the target cell genome has shown promise as an affordable option for gene therapy.13 To date, CRISPR clinical trials illustrate efficacy in LCA-10, rhodopsin mediated retinitis pigmentosa, and neovascular (i.e., “wet”) age-related macular degeneration.13
Conclusions
With the advent of potential new treatments, gene therapy has made remarkable strides in the management of IRDs in a relatively short time period. As described above, gene therapy technology contains several approaches addressing a variety of conditions. As genetic testing continues to mature and become economically feasible, it will undoubtedly become a clinically indispensable tool to improve the accuracy of IRD diagnosis.
As these emerging gene therapies become approved, ophthalmologists need to remain educated and abreast of the underlying disease pathophysiology to best serve the rapidly changing clinical needs.
References
- Bessant DA, Ali RR, Bhattacharya SS. Molecular genetics and prospects for therapy of the inherited retinal dystrophies. Current opinion in genetics & development. 2001 Jun 1;11(3):307-16.
- RetNet: Disease Table. (n.d.). Retrieved April 18, 2021, from https://sph.uth.edu/retnet/disease.htm
- Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388(10045):661-672. doi:10.1016/S0140-6736(16)30371-3
- Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018 Jan 12;359(6372).
- Genetic Disorders. (n.d.). Retrieved April 18, 2021, from https://www.genome.gov/For-Patients-and-Families/Genetic-Disorders
- Bundey SA, Crews SJ. A study of retinitis pigmentosa in the City of Birmingham. I Prevalence. Journal of medical genetics. 1984 Dec 1;21(6):417-20.
- Takahashi VK, Takiuti JT, Jauregui R, Tsang SH. Gene therapy in inherited retinal degenerative diseases, a review. Ophthalmic genetics. 2018 Sep 3;39(5):560-8.
- Garafalo AV, Cideciyan AV, Héon E, Sheplock R, Pearson A, Yu CW, Sumaroka A, Aguirre GD, Jacobson SG. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Progress in retinal and eye research. 2019 Dec 30:100827.
- Rodrigues GA, Shalaev E, Karami TK, Cunningham J, Slater NK, Rivers HM. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharmaceutical research. 2019 Feb;36(2):1-20.
- Gupta PR, Huckfeldt RM. Gene therapy for inherited retinal degenerations: initial successes and future challenges. Journal of neural engineering. 2017 Aug 22;14(5):051002.
- Russell SR, Bennett J, Wellman J, Chung DC, High K, Yu ZF, Tillman A, Maguire AM. Three-year update for the phase 3 voretigene neparvovec study in biallelic RPE65 mutation–associated inherited retinal disease. Investigative Ophthalmology & Visual Science. 2018 Jul 13;59(9):3900-
- Prado DA, Acosta-Acero M, Maldonado RS. Gene therapy beyond luxturna: a new horizon of the treatment for inherited retinal disease. Current opinion in ophthalmology. 2020 May 1;31(3):147-54.
- Peddle CF, Fry LE, McClements ME, MacLaren RE. CRISPR Interference–Potential Application in Retinal Disease. International journal of molecular sciences. 2020 Jan;21(7):2329.
