




Pigmented fundus lesions are commonly encountered on routine ophthalmological dilated examinations and may be sent to your practice for further evaluation after being identified by another health care professional. Pigmented lesions may represent a benign or malignant process. They can be congenital, acquired, or the result of an infectious or inflammatory process affecting the retinal pigment epithelium (RPE), the layer that provides the natural pigmentation of the fundus.
Generally, outside of congenital lesions, pigmented lesions tend to be seen more in the elderly population (over 40), with a split prevalence among males and females. Prevalence rates vary from 0.2 to 30% for choroidal nevi.1-3
Some lesions have discrete focal patterns, while others may be more diffuse; various lesions have a predisposition for specific ethnic groups, and others may occur due to different environmental exposures. Sometimes a combination of factors may lead to the development of a pigmented lesion.
RPE cells contain melanosomes of neuroectodermal origin that develop during the second trimester of development. RPE cells transport nutrients, ions, and water, phagocytose the outer rods and cones, maintain the blood-ocular border, metabolize vitamin A, secrete factors essential to the integrity of the retina structurally, convert all-trans-retinal into 11-cis retinal, and absorb light with protection against photooxidation.4
Disruptions of the RPE layer may lead to:
- hypertrophy
- hyperplasia
- migration
- metaplasia
- atrophy of RPE cells, producing various pigmented configurations.4
Most pigmented lesions are asymptomatic and discovered incidentally during routine eye exams; however, some may present with symptoms ranging from floaters, field loss, photopsias, reduced visual acuity, and pain. While some lesions may be harmless, others may be sight or even life-threatening.
Distinguishing and diagnosing lesions represents a diagnostic challenge for eyecare providers. Some studies reveal as low as a third of new intraocular tumor patients referred to tertiary ocular oncology care centers are sent with the correct diagnosis.5 In this overview, we will discuss the differential diagnoses and the management of some of the more common pigmented lesions encountered on routine clinical examination.

Download the Pigmented Fundus Lesion Cheat Sheet
This comprehensive cheat sheet serves as a differential diagnosis guide for pigmented fundus lesions.

Congenital Hypertrophy of Retinal Pigment Epithelium (CHRPE)
Congenital hypertrophy of retinal pigment epithelium (CHRPE) represents an asymptomatic solitary, flat, and well-demarcated pigmented lesion whose shape may be round, elliptical, or irregular. These lesions may also contain non-pigmented areas within called "lacunae" and non-pigmented lines at the lesional margins ("halo"). Additionally, there may be overlying retinal vessel abnormalities such as vascular sheathing and attenuation.
As the name suggests, CHRPE occurs secondary to hypertrophy and hyperpigmentation of the RPE layer. CHRPE lesions should be classified as typical or atypical.
Typical lesions are often round and solitary as described above and may even feature enlargement of the lacunae over time. Typical CHRPE lesions can sometimes have a "bear track" appearance when multiple small lesions are seen in a cluster pattern.
Atypical lesions include pigmented RPE lesions that are pisciform or spindle-shaped, bilateral, and multiple in number. Atypical presentations of CHRPE may represent a higher predisposition for familial adenomatous polyposis (FAP), a risk factor for developing colon cancer.
Figure 1 demonstrates typical grouped CHRPE.

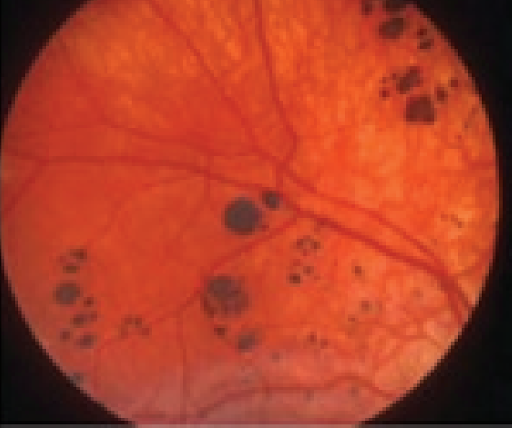
Figure 1: Mishra et al.
Figure 2 illustrates atypical pisiform CHRPE.

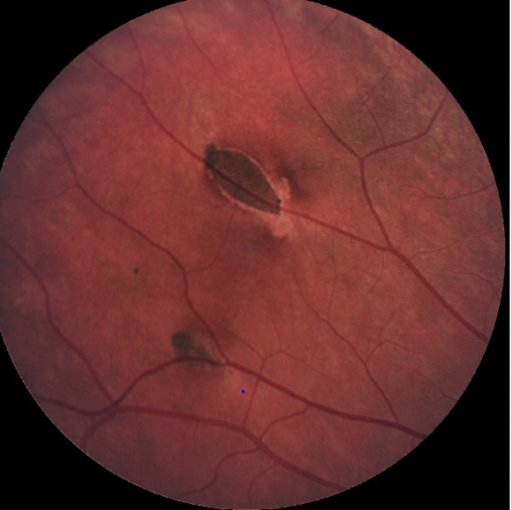
Figure 2: Diebert et al.
On fundus autofluorescence (AF), CHRPE displays a uniform area of hypo-autofluorescence while the lacunae appear iso-auto fluorescent. On infrared (IR) imaging, CHRPE lesions are hyporeflective, while the lacunae are hyperreflective.

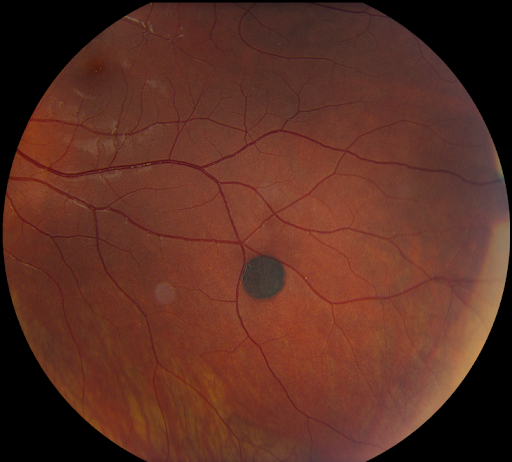
Figure 3
On OCT, CHRPE appears as a thicker, brighter RPE band with overlying disruption (sometimes absence) of the outer nuclear layer and ellipsoid zone (see Figure 4 below).

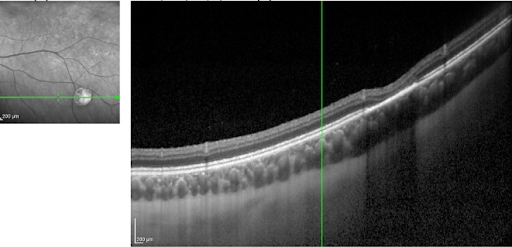
Figure 4
On B-Scan, CHRPE appears flat with high internal reflectivity (see Figure 5 below).

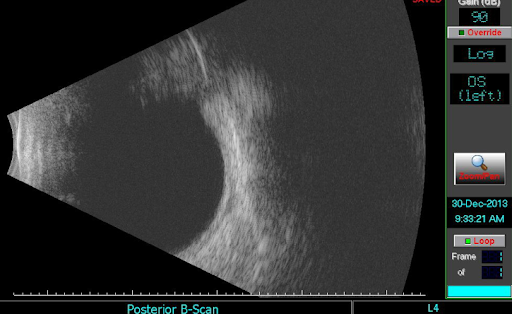
Figure 5

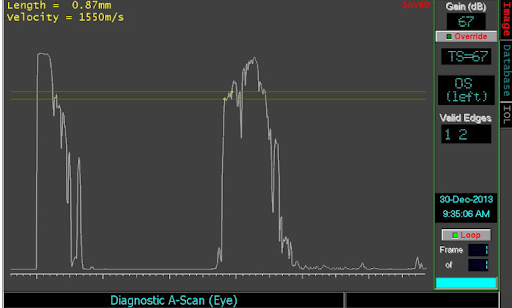
Figure 6: Diagnostic A scan
Differentials of CHRPE include choroidal nevi, choroidal melanoma, toxoplasmosis, and lattice degeneration.
RPE Hyperplasia
RPE hyperplasia occurs when there is a non-specific proliferation of RPE cells that forms an irregular-shaped pigmented retinal lesion sometimes secondary to inflammation, trauma, vitreous traction, or therapeutic interventions.
In Figure 7, RPE hyperplasia is represented.

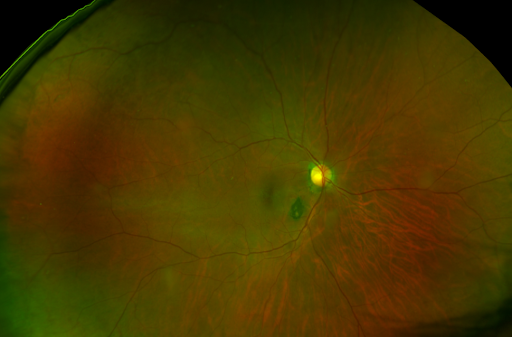
Figure 7: Daytona
Often smaller areas of RPE hyperplasia are described as "pigment clumping." Pigment clumping in the macula (see Figure 8 below) is one of several characteristics of dry age-related macular degeneration discussed later in this paper.
Figure 8 demonstrates RPE pigment clumps in the macula (bilateral).

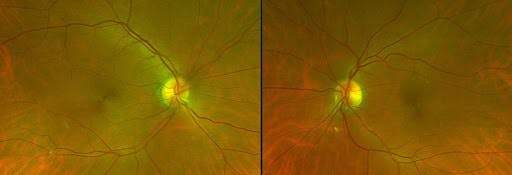
Figure 8
Choroidal Nevi
Choroidal nevi are benign, often asymptomatic pigmented lesions typically round with well-defined borders. Reported incidence has been highly variable from 0.2 to 30%, with higher rates reported in lighter pigmented individuals.3
Typically, nevi are without subretinal fluid or orange pigment, do not juxtapose the optic nerve, include overlying drusen, and are no thicker than 1.0 mm. Atypical nevi may have ill-defined borders and appear less uniform with less pigmentation. Nevi should be serially documented and objectively photographed and measured with B-Scan ultrasound to monitor features such as size (i.e., diameter and depth) and internal reflectivity over time.
Progression in size, new development of subretinal fluid or orange pigment should prompt a re-evaluation of the diagnosis. Malignant transformation of choroidal nevi to melanoma is rare, with estimates as low as 1 of every 8000.6 Other imaging modalities may help diagnose nevi as well, including fluorescein angiography in which nevi remains hyperfluorescent, or OCT in which nevi may be associated with RPE atrophy, PR loss and IS/OS irregularity. On FAF, the nevus appears hypo autofluorescent.
Figure 9 of a typical choroidal nevus.

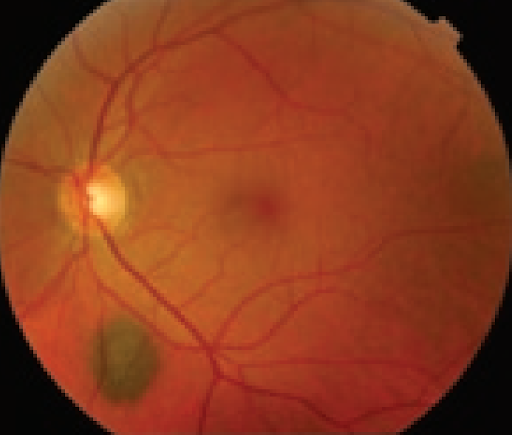
Figure 9
In Figure 10, notice typical choroidal nevi with small drusen.

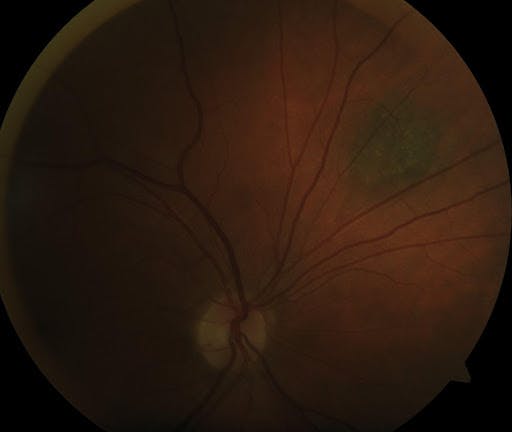
Figure 10
In Figure 11, OCT with enhanced depth imaging is able to highlight the posterior edge of the choroidal lesion.

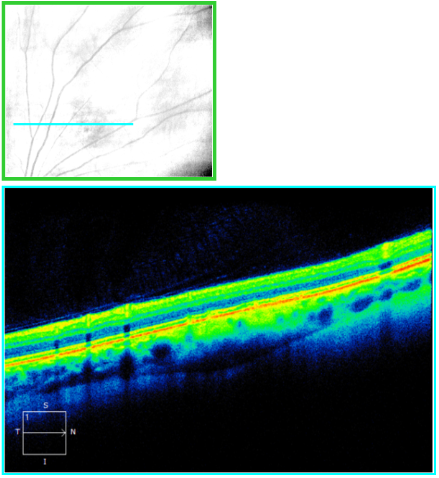
Figure 11
Refer to Figure 12 for atypical nevi with irregular borders and lighter pigmentation.

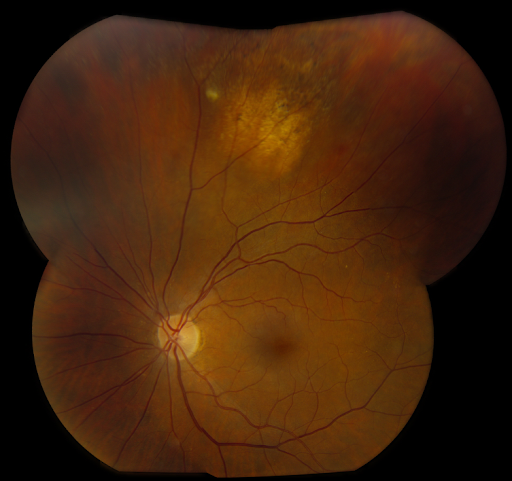
Figure 12
Choroidal melanoma
Choroidal melanoma is the most common primary intraocular malignancy in adults and the second most common intraocular tumor overall (after metastasis).3 They can present with photopsia, floaters, decreased vision, and visual field loss. In nearly a third of patients, choroidal melanoma is detected on routine screening before symptoms develop. Choroidal melanoma can sometimes be challenging to distinguish from nevi, especially when small in size; however, they are typically greater in thickness (>2.0 mm) and larger in diameter (> 4 disc diameters).
Other key features of choroidal melanoma include orange pigment, associated subretinal fluid, and growth over time. Larger choroidal melanomas have a distinct mushroom or dome shape and often have a concomitant exudative retinal detachment. Diffuse melanomas that are not as thick but have a broad base are often aggressive with extraocular extension. Dangerous characteristics can be represented by the "MOLES" system.
"MOLES" system for classification of choroidal melanoma:
- M-mushroom shape
- O-orange pigment
- L-large size
- E-enlarging, and
- S-subretinal fluid
A MOLES score of greater than 2 represents a high sensitivity metric for malignancy. Genetic analysis and close observation are options in cases where diagnosis is equivocal. Treatment depends on the stage and grade of the tumor and is often multi-disciplinary, including brachytherapy, radiotherapy, laser therapy, enucleation, and surgical resection. The risk of recurrence and long-term complications from retinal detachment, macular edema, and neovascularization requires continued long-term surveillance
Figure 13 demonstrates choroidal melanoma.

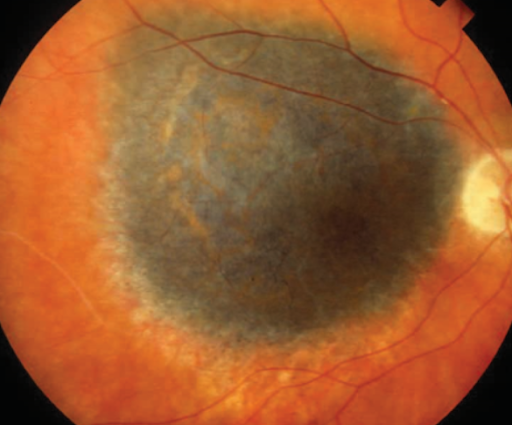
Figure 13: Griggs P, AAO.
Figure 14 is a montage photo of choroidal melanoma.

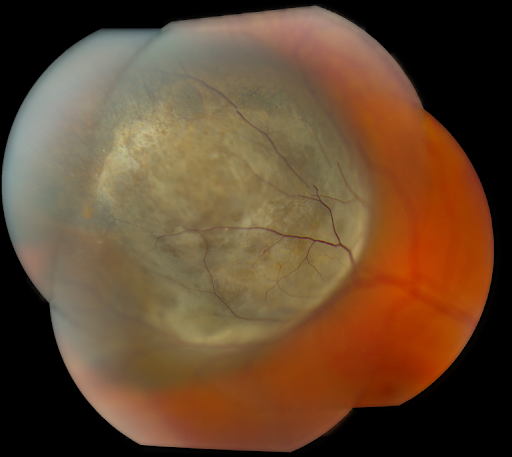
Figure 14
Lattice degeneration
Lattice degeneration represents a group of frosty-appearing granules found in nearly 10% of the general population and almost half of the individuals with myopic eyes.1 Lattice degeneration is sometimes associated with retinal thinning, vitreous liquefaction, and vitreoretinal traction. Lattice degeneration may lead to retinal tears in up to 54% of patients, and nearly a third of retinal detachment patients have lattice degeneration.7
Lattice degeneration is also seen with hereditary vitreoretinopathies, such as Stickler and Wagner syndromes. The exact etiology is unclear and various theories exist, including developmental anomalies of the internal limiting membrane and secondary to retinal ischemia. The diagnosis can be made clinically, and on OCT imaging, there may be irregular surfaces and vitreoretinal adhesions with or without traction.
Differential diagnoses include pavingstone degenerations, retinoschisis, atrophic holes, CHRPE, chorioretinal scarring, and white without pressure. Prophylactic barricade laser is sometimes indicated, especially if an associated retinal hole, tear, or detachment is associated.
Figure 15 offers an example of lattice degeneration.

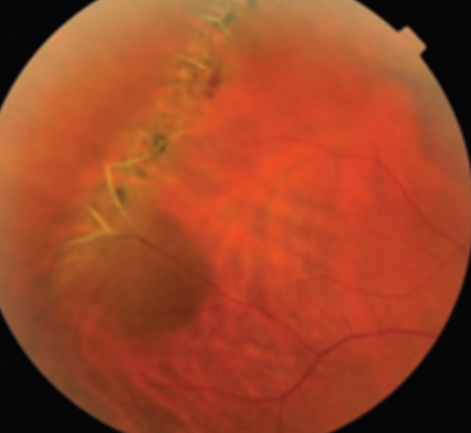
Figure 15: ASRS
Melanocytoma
Melanocytomas are typically unilateral black lesions that commonly occur over the optic nerve and may extend into the choroid or neurosensory retina. They occur at similar frequencies across various ethnic groups and are slightly more prevalent in females. Melanocytomas are benign with a good prognosis. Severe visual disturbances are uncommon, but a quarter of patients may experience a mild degree of vision loss. Etiologies responsible for severe manifestations in the setting of melanocytoma include central retinal vein occlusions or malignant transformation.
Growth itself does not necessarily indicate malignant transformation as up to 15% of melanocytomas demonstrate enlargement over the years.8 The highest risk factor for malignant transformation is a thickness at a 1.5 mm or greater presentation.8 Visual field defects and afferent pupil defects are common, up to 90% and 30%, respectively.9 Due to the risk of malignant transformation in 1-2% of cases, observation is recommended with serial fundus photography.
An important differential diagnosis is uveal melanoma; however, uveal melanoma tends to occur more in lighter pigmented individuals while melanocytoma occurs equally in all ethnic groups. Other diagnoses to consider include optic nerve glioma, capillary hemangioma, and metastasis.
While the diagnosis is mainly clinical, imaging such as OCT of the retinal nerve fiber layer (RNFL) or B-scan ultrasound can be utilized to assess the risk of malignancy and to differentiate melanocytoma from other more sinister lesions. On B-scan ultrasound low internal reflectivity typically indicates a benign lesion.
Metastatic melanoma of the optic disk tends to grow more rapidly and diffusely, while choroidal melanoma invades the sensory retina. Imaging, as mentioned above, helps differentiate these lesions.
Figure 16 illustrates melanocytoma.

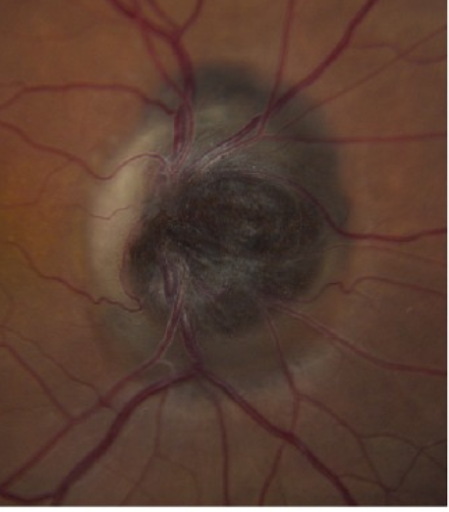
Figure 16: De Alba MA et al.
In Figure 17, small melanocytoma at the temporal disc with peripapillary laser scarring is represented.

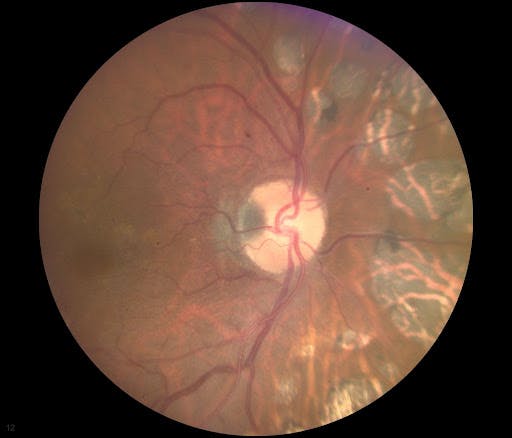
Figure 17
Choroidal rupture
Choroidal rupture represents a break in the choriocapillaris, Bruch membrane, and RPE with an intact overlying neurosensory retina, usually occurring in the setting of trauma and closed globe injury. After the initial insult and hemorrhage clears, choroidal neovascularization (CNV) can develop with or without serous or hemorrhagic pigment epithelial detachments. Diagnosis made from a combination of history of closed globe injury with a crescent-shaped yellow/white subretinal streak that generally is near the disc.
FA will show hypofluorescence early and late staining and late leakage if CNV is present. Autofluorescence will reveal hypofluorescence at the site of rupture with hyperfluorescence at the border edge of the rupture. OCT shows disruption of the RPE at the site of the rupture, sometimes with a thin underlying choroid. Symptoms depend on the rupture location, with metamorphopsia or decreased vision when occurring in the fovea. These lesions are typically observed with Amsler monitoring.
Anti-VEGF therapy may be indicated if there is development of CNV Differential diagnosis of choroidal rupture include lacquer cracks in patients with high myopia and angioid streaks that emanate from the disc and are also associated with CNV, however, these are both often bilateral and do not typically have an associated history of trauma.
Refer to Figure 18, below, for an image of choroidal rupture (nasal macula) and choroidal nevi (inferonasal to nerve).

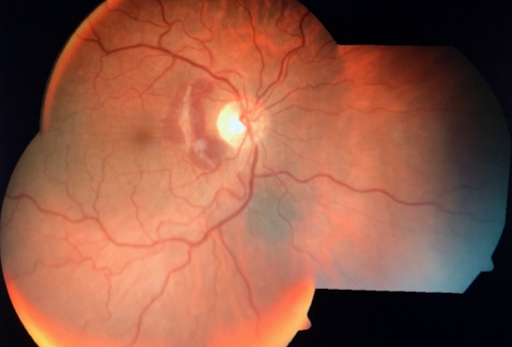
Figure 18: Eyewiki
Figure 19 demonstrates a traumatic choroidal rupture with choroidal neovascularization involving the fovea.

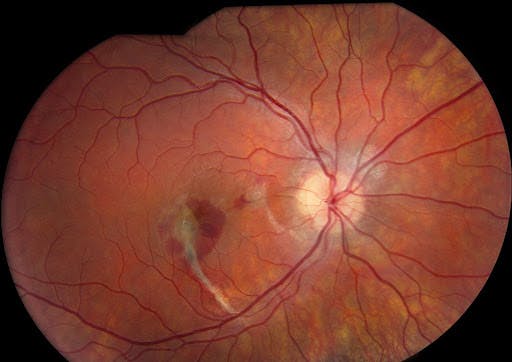
Figure 19
Toxoplasmosis
Toxoplasmosis classically presents as focal retinitis with overlying vitreous inflammation, described as "headlights in a fog," with a nearby pigmented chorioretinal scar. The differential for toxoplasmosis includes other infectious processes, including tuberculosis, toxocariasis, syphilis, cytomegalovirus, herpes simplex virus, varicella-zoster virus, and autoimmune processes, such as Behcet's disease and retinal vasculitis.
Lesions threatening the macula or optic nerve, causing visual impairment during pregnancy or in an immunocompromised host represent situations where treatment is indicated.
Treatment includes systemic antibiotics with trimethoprim/sulfamethoxazole (Bactrim), azithromycin, or clindamycin. Intravitreal antibiotics can also be considered. Outcomes largely depend on the location of the lesions and degree of inflammation. Lesions can relapse and reoccur; however, bilateral impairment is rare.
Figure 20 depicts a congenital toxoplasmosis scar.

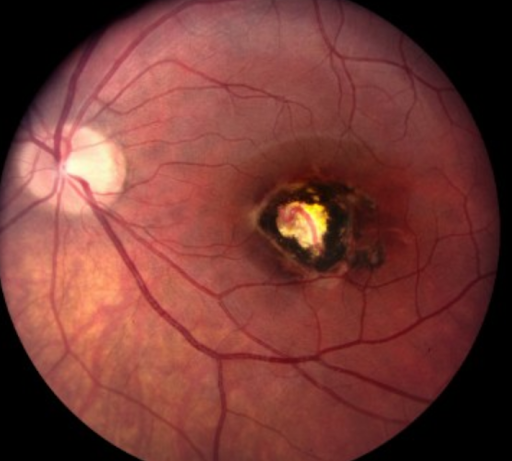
Figure 20: AAPOS
Figure 21 illustrates a toxoplasmosis scar with active chorioretinitis.

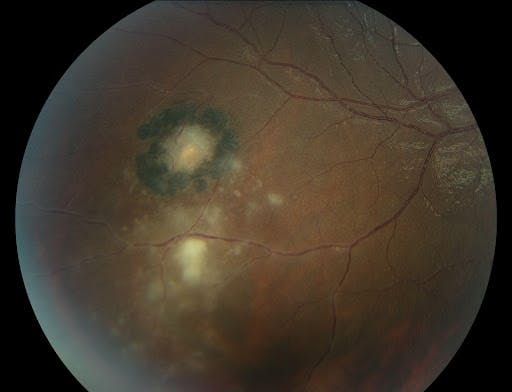
Figure 21
Metastatic carcinoma
Metastatic processes can occur in any part of the eye, including the eyelid, conjunctiva, or orbit, and are the most common intraocular tumor. Whenever coming across a new pigmented lesion involving the choroid or retina, a thorough history of any prior history of malignancy should be reviewed, even if the malignancy is said to be in remission.
Breast cancer represents the most common primary tumor that can metastasize to the eye; however, metastasis can occur secondary to colon, renal, prostate, or skin tumors.10 The majority of patients with breast cancer metastasis have a known diagnosis of cancer; however, this rate is not as high as other cancers such as lung metastasis, where only around 66% of patients have a cancer diagnosis.11
When there is sufficient concern for metastasis to the eye without a known history of malignancy, further investigation should be done by the aid of the primary team (e.g., chest imaging, liver function tests, mammograms, colonoscopy). Metastasis from lung cancer is also unique in that they are rarely bilateral and are sometimes painful. It is also vital to differentiate metastasis from primary melanomas. Breast cancer metastases, for example, tend to be smaller (<3 mm) compared to the average thickness of choroidal melanomas (5.5 mm).10
Metastasis are also unique in appearance in that they tend to be yellow, homogenous and the overlying RPE is not disrupted to the extent in melanoma. Although creamy yellow lesions are classic, metastasis may also present with exudative retinal detachments or optic nerve edema.
Any lesion suspected to be ocular metastasis should be referred to an ocular oncologist. Systemic disease will be treated in conjunction with the primary oncology team.
In Figure 22, ocular metastasis from lung cancer is pictured.

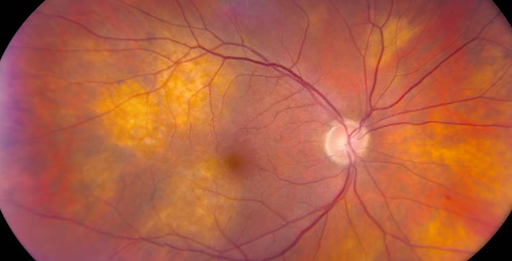
Figure 22: University of Iowa
Figure 23, below, shows multifocal choroidal metastasis from neuroendocrine tumor.

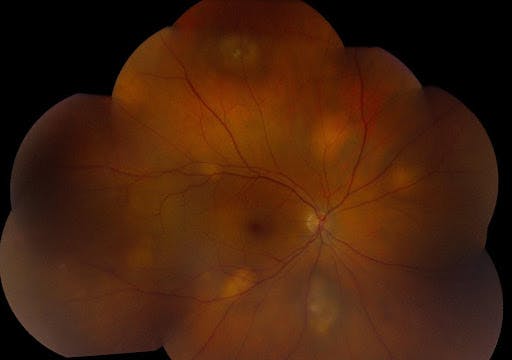
Figure 23
Figure 24 represents uterine cancer with choroidal metastasis.

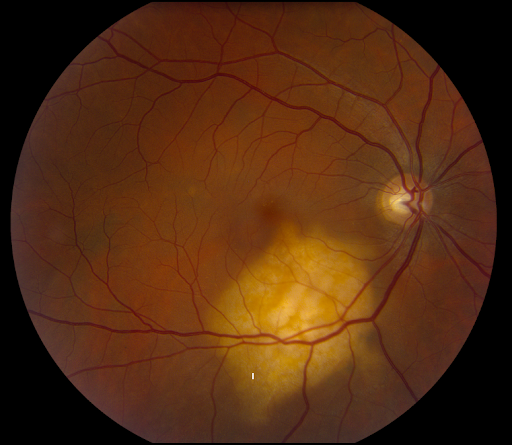
Figure 24
Coloboma
Colobomas represent a failure or incomplete closure of the choroidal or optic fissure during development. They can occur in the iris, ciliary body, choroid, retina, and optic nerve. Colobomas of the choroid, retina, or optic nerve result from failure of posterior closure. The incidence of ocular colobomas is rare, occurring in 0.5 to 2.2 cases per 10,000 live births.12 Colobomas can be associated with many systemic disorders and other eye abnormalities.
Individuals with posterior colobomas have a higher risk of retinal detachment (some estimates stating as high as 40%) and typically appear as an area of whitening with pigment deposition at the border of normal retina and the coloboma.12
Patients with bilateral colobomas are sometimes referred to a genetics specialist to evaluate for systemic disorders. Typically colobomas are observed; however, some may be barricaded with prophylactic laser to reduce the risk of retinal detachment. Other diagnoses to consider when colobomas occur posteriorly include optic nerve pits, optic nerve staphylomas, morning glory discs, and other general inflammatory lesions.
In Figure 25, coloboma involving optic nerve, retina, and choroid is present.

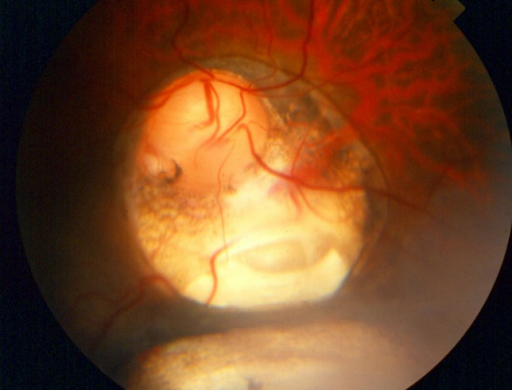
Figure 25: AAO
Age-related macular degeneration
Age-related macular degeneration (AMD) is the leading cause of central vision loss in the elderly in Western nations.13 AMD and its respective stages change the appearance of the macula in various ways. Signs include drusen, RPE changes, subretinal fibrosis, geographic atrophy, and subretinal fluid. Dry AMD does not typically cause severe vision loss, while wet AMD is a significant cause of vision loss.
Dry and wet AMD can lead to distorted near vision, scotoma, metamorphopsia, and micropsia. FA and OCT help evaluate the presence of wet AMD. OCT should be used to monitor the patient during follow-up visits, and FA should be utilized when suspicion develops for CNVM.
The differential diagnosis for wet AMD includes other choroidal neovascularization causes, including pathologic myopia, angioid streaks, choroidal rupture, and ocular histoplasmosis. Other foveal pathology such as pigment epithelial detachments or vitelliform lesions will not typically leak on FA. Pachychoroid spectrum disorders such as polypoidal choroidal vasculopathy may develop pigment epithelial detachments; however, these detachments are not associated with drusen. Management for dry AMD includes AREDS2 vitamins, smoking cessation, and Amsler grid monitoring. Anti-VEGF therapy is indicated if there are signs of wet AMD.
Conclusion
A systematic approach is needed when diagnosing and working up pigmented fundus lesions. It is important to characterize critical aspects of the lesion, such as the anatomical level of the lesion, color, size/depth, shape, surface features, laterality, etc. Imaging modalities such as B-Scan ultrasound, OCT imaging, color fundus photographs, and fluorescein angiography may aid in initially and subsequently evaluating such lesions. Monitoring these lesions is critical as changes may alter the initial diagnosis and affect prognosis.
Download this cheat sheet as a differential diagnosis guide for pigmented fundus lesions.
References
- Pickering M, Luciani L, Zaour N, Petrella R. Prevalence, incidence, and characteristics of patients with choroidal neovascularization secondary to pathologic myopia in a representative Canadian cohort. Invest. Ophthalmol. Vis. Sci. 2014;55(13):3621.
- Silvestri G, Williams MA, McAuley C, et al. Drusen prevalence and pigmentary changes in Caucasians aged 18-54 years. Eye (Lond). 2012;26(10):1357-1362. doi:10.1038/eye.2012.165
- Nicola B Quinn, David Wright, Tunde Peto, Sharon Mary Cruise, Ian Young, Frank Kee, Usha Chakravarthy, Ruth E Hogg; Prevalence and characteristics of peripheral retinal lesions in an aging population.. Invest. Ophthalmol. Vis. Sci. 2017;58(8):1497.
- Rafael Simó, Marta Villarroel, Lídia Corraliza, Cristina Hernández, Marta Garcia-Ramírez, "The Retinal Pigment Epithelium: Something More than a Constituent of the Blood-Retinal Barrier—Implications for the Pathogenesis of Diabetic Retinopathy", BioMed Research International, vol. 2010, Article ID 190724, 15 pages, 2010. https://doi.org/10.1155/2010/190724
- Law C, Krema H, Simpson ER. Referral patterns of intraocular tumour patients to a dedicated Canadian ocular oncology department. Can J Ophthalmol 2012;47:254Y61.
- Singh AD, Kalyani P, Topham A. Estimating the risk of malignant transformation of a choroidal nevus. Ophthalmology. 2005 Oct;112(10):1784-9. doi: 10.1016/j.ophtha.2005.06.011. PMID: 16154197.
- Byer NE. Lattice degeneration of the retina. Surv Ophthalmol. Jan-Feb 1979;23(4):213-48.
- Shields JA, Demirci H, Mashayekhi A, et al. Melanocytoma of the optic disc: a review. Surv Ophthalmol. 2006;51:93–104.
- Al-Rashaed S, Abboud EB, Nowilaty SR. Characteristics of optic disc melanocytomas presenting with visual dysfunction. Middle East Afr J Ophthalmol. 2010;17:242-5.
- Ilias Georgalas, Theodore Paraskevopoulos, Chryssanthi Koutsandrea, Evgenia Kardara, Panagiotis Malamos, Dimitrios Ladas, Dimitris Papaconstantinou, "Ophthalmic Metastasis of Breast Cancer and Ocular Side Effects from Breast Cancer Treatment and Management: Mini Review", BioMed Research International, vol. 2015, Article ID 574086, 8 pages, 2015. https://doi.org/10.1155/2015/574086
- Lampaki S, Kioumis I, Pitsiou G, et al. Lung cancer and eye metastases. Med Hypothesis Discov Innov Ophthalmol. 2014;3(2):40-44.
- Nakamura KM, Diehl NN, Mohney BG. Incidence, ocular findings, and systemic associations of ocular coloboma: a population-based study. Arch Ophthalmol. 2011;129(1):69-74. doi:10.1001/archophthalmol.2010.320
- Harvey PT. Common eye diseases of elderly people: identifying and treating causes of vision loss. Gerontology. 2003 Jan-Feb;49(1):1-11. doi: 10.1159/000066507. PMID: 12457044.
