




In 2021, 3,320 new cancers of the eye and orbit were diagnosed, 400 of which led to death in the United States.1 Ocular tumors represent a diverse group of tumors that can affect multiple layers of the eye. For any given tumor, there exists predispositions for certain ethnic and age groups. In addition to demographic and environmental risk factors, certain tumors have a genetic association and predisposition. There has been a further classification of genetics role in diagnosis, prognostication, and predictive modeling for ocular tumors in recent years.
Tumors of the eye can be “primary” and originate from within the eye. Or, more commonly, they can be the “secondary” type which can occur as metastasis or expansion of a primary tumor originating from elsewhere in the body. In primary tumors, the most common intraocular tumor in adults is uveal melanoma (iris, choroid, or ciliary body), followed by primary intraocular or vitreoretinal lymphoma. In contrast, in children, the most common is retinoblastoma.1,2
In this article, we will focus on the most common ocular tumors and the recent advances of genetics in each condition.
Ocular melanoma
Ocular melanoma includes posterior uveal (choroidal) melanoma, iris melanoma, and conjunctival melanoma, with all having distinct genetic profiles. Ocular melanoma represents the second most common type of melanoma after skin melanoma.2 Within the different types of ocular melanoma, uveal represents the most common, with a worldwide incidence between 5 to 9 cases per million.2
Uveal melanoma
Uveal melanoma most commonly occurs in the choroid, followed by the ciliary body and iris (in the pediatric population, the incidence of iris melanoma is significantly higher). In terms of survival rate, the literature suggests a 5-year overall survival rate of nearly 80% for uveal melanomas; however, mortality has been noted to be as high as 50% when metastasis is present.2 Treatment includes surgery, radiotherapy, or a combination thereof. These treatment modalities have limited long-term survival, and there is no definitive treatment for metastatic uveal melanoma.
Genetics play a factor in prognosis and treatment modalities. Proto-oncogenes demonstrate high relevance as activation of the MAPK and GNAQ pathways has been observed in around 50% of uveal melanomas.3,4 As such, immunotherapy targeting the GNAQ and GNA11 genes and the MAPK-Erk pathway utilizing anti-PD-1 monoclonal antibodies such as pembrolizumab have been pursued and shown to be effective in a minority of patients.3,4
Beyond being a target for treatment, genetic and cytogenetic factors also impact prognosis and can be correlated with tumor diameter, cell type, mitotic or Ki-67 indexes and ciliary body involvement to fully prognosticate.
While most uveal melanomas are thought to be sporadic, in recent years, genetic patterns of uveal melanomas have been studied, with emerging data and research suggesting family clustering and the discovery of somatic mutations in genes such as BAP1 that may provide further insight into pathogenesis and lead to the ability to perform genetic screening.3,4 At the minimum, BAP1 plays a role in a subset of hereditary uveal melanomas.
Low HLA class I expression is associated with a decreased risk of metastasis.3,4 As such, HLA has also been an area of focus as natural killer cells target cells with low HLA expression, making them a key player in targeting tumor cells. Additionally, HLA-B40 is associated with metastatic death from UM. Lastly, individuals with ligands for both KIR2DLI and KIR2Dl2/3 rather than individually have been shown to have better outcomes, providing yet more momentum for targeting immunotherapeutic strategies.3,4
Genetic mutations in cutaneous melanoma have also been examined in the context of uveal melanoma. Mutations in MCR1 and CDKN2A, for example, have shown to increase the risk of cutaneous melanoma; however, these mutations and others commonly implicated in cutaneous melanoma have not also demonstrated susceptibility to uveal melanoma.3,4
The focus of current research is identifying and examining susceptibility genes that play a role in the development of uveal melanoma. Particular attention on families with strong history, and on those with other conditions such as cutaneous melanoma. At present, the identification of germline mutations is limited. However, some areas of interest have been identified, particularly surrounding HLA, Natural Killer cells, and BAP1.
Uveal melanoma represents an arena where genetic markers significantly advance prognosis. Luo H et al. recently uncovered 21 genes that can be used to accurately identify patient prognosis to an individual level (i.e., SERPINB9, EDNRB, RAPGEF3, HFE, RNF43, ZNF415, IL12RB2, MTUS1, NEDD9, ZNF667, AZGP1, WARS, GEM, RAB31, CALHM2, CA12, MYEOV, CELF2, SLCO5A1, ISM1, and PAPSS2).3
Additionally, newer genetic testing techniques such as multiplex ligation-dependent probe amplification and gene expression profiling have built upon karyotype analysis, fluorescein in site hybridization, and comparative genomic hybridization and can prognosticate the risk of metastasis in uveal melanoma.
Chromosomal abnormalities have also recently become a source for prognostication, for example, monosomy 3. The presence of monosomy 3 has been associated with a poor prognosis and greater risk of metastasis as compared to those individuals without such an abnormality in chromosome 3 to the extent that the majority of tumors with monosomy 3 develop metastasis while less than 5% of tumors without monosomy 3 have been shown to develop metastasis.5 DecisionDx-UM has developed a screening tool that has become a standard of practice for uveal melanoma to risk-stratify patients for metastasis.6
Below, Figure 1 illustrates primary choroidal melanoma.

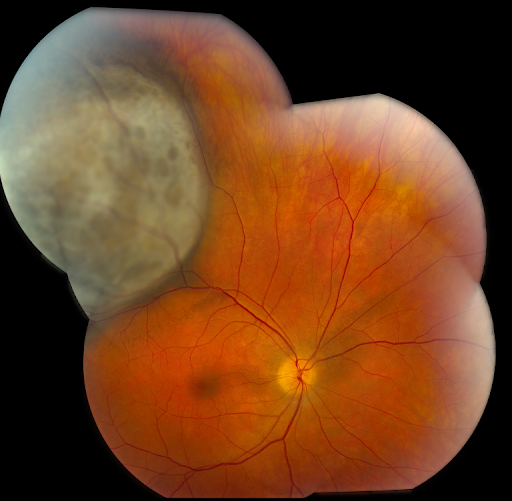
Figure 1
Figure 2 demonstrates a partially amelanotic lesion in the iris.

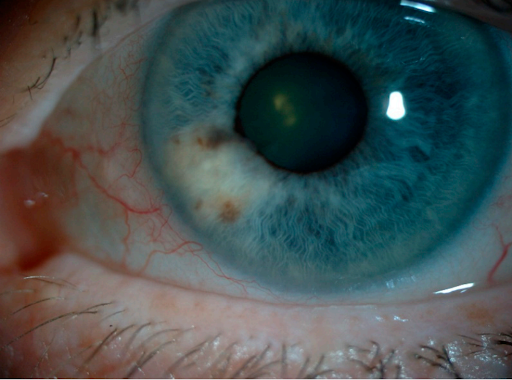
Figure 2: Photo Credit Van Poppelen et al (2021)
Primary intraocular lymphoma/vitreoretinal lymphoma
Vitreoretinal lymphoma is a subtype of primary central nervous system lymphoma and is the most common intraocular lymphoma and affects the vitreous, retina, and optic nerve.7 The majority of primary vitreoretinal lymphoma are non-Hodgkin's diffuse B cell lymphomas (DLBCL). The two major subtypes of DLBCL are germinal center B cell (GCB) and activated B cell (ABC).
Patients with the ABC subtype have a far poorer prognosis than GCB lymphomas. Emerging literature has reported genetic variations in chromosome 6p21.32 being associated with susceptibility with DLBCL, and chromosomal translocation of BCL6 at 3q27 has been identified as the most characteristic genetic abnormality seen in DLBCL.7,8
Infectious agents such as DNA from EBV, HHV8, and toxoplasmosis may play a role in the development of vitreoretinal lymphoma in immunocompromised patients. Additionally, elevated levels of IL-10 mRNA in intraocular or CSF have been highly correlated with B cell vitreoretinal lymphomas.
A new diagnostic development involving genetics in the case of vitreoretinal lymphoma involves anterior chamber taps of aqueous humor. Hiemcke-Jiwa LS et al. found in 63 patients studied that there was nearly a 90% concordance between small volume aqueous samples and vitreous fluid samples for the MYD88 L265P mutation.7 This provides a promising and minimally invasive method to diagnose accurately, detect recurrence, and monitor treatment.
Retinoblastoma
Retinoblastoma represents the most common intraocular malignancy in children, with an incidence of 1 in 15,000 births.9.10 The average age of diagnosis is 18 months, with unilateral cases being 24 months and bilateral earlier than 12 months. Retinoblastoma does not have an ethnic or gender predisposition. Retinoblastoma is caused by germline or somatic inactivation of the susceptibility gene RB1 (RB is inheritable in 45% of cases).9,10
Genetic testing can provide further insight as to whether or not the initial RB1 mutation detected is germline or somatic in origin. This distinction carries weight as the hereditary form carries an increased risk of developing secondary malignancies. Techniques in practice allow for detection of the RB1 mutation in 97% of cases with a known family history or those without family history but with bilateral or multiple unilateral tumors.
In terms of treatment, there has been a relative shift from preserving the eye to preserving the vision by more precise and earlier identification of the RB1 gene mutation. One treatment modality focused on activating p53 induced cell death of RB cells by interfering with MDMX p53 and MDM2 p53 interactions has shown promising results in killed RB cells in mouse models.9.10
Epigenetics has also been an area of focus in retinoblastoma. That is, the proto-oncogene SYK is upregulated and is key for tumor cell survival. Targeting SYK with small molecule inhibitors may markedly induce RB tumor cell growth.9.10
Figure 3 shows a pediatric patient with retinoblastoma.

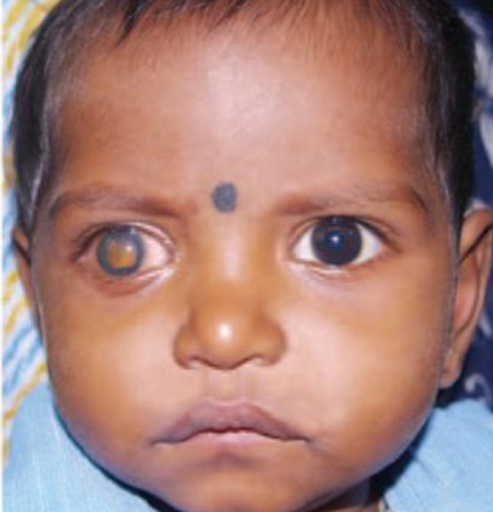
Figure 3: Nagarkatti-Gude et al 2012
With all of the above tumors and others, epigenetics have also become an area of increasing focus in the context of ocular tumors due to the fact that epigenetic changes are reversible. New medications such as the 5-Aza have been shown to decrease the number of metastases from the eye to the lung in marine models while HDAC inhibitors may prolong dormancy of micrometastatic disease.9,10
Conclusions
Genetic tests have the potential to examine the individual inherited risk of developing specific cancers. The presence of genetic mutations may predict risk; however, their presence does not always directly translate to the occurrence of disease, and the absence of genetic markers does not always absolve an individual from developing a given disease. Combining genetics with clinical presentation and potential confounding environment variables is critical.
References
- American Cancer Society. Cancer Facts & Figures 2021. Atlanta, Ga: American Cancer Society; 2021.
- Van Poppelen NM, de Bruyn DP, Bicer T, et al. Genetics of Ocular Melanoma: Insights into Genetics, Inheritance and Testing. Int. J. Mol. Sci. 2021, 22, 336.
- Luo H, Ma C (2020) Identification of prognostic genes in uveal melanoma microenvironment. PLOS ONE 15(11): e0242263.
- Helgadottir H, Höiom V. The genetics of uveal melanoma: current insights. Appl Clin Genet. 2016;9:147-155. Published 2016 Sep 6. doi:10.2147/TACG.S69210
- Maria T. Sandinha, Maura A. Farquharson, Ian C. McKay, Fiona Roberts; Monosomy 3 Predicts Death but Not Time until Death in Choroidal Melanoma. Invest. Ophthalmol. Vis. Sci. 2005;46(10):3497-3501. doi: https://doi.org/10.1167/iovs.05-0613.
- Harbour JW, Chen R. The DecisionDx-UM Gene Expression Profile Test Provides Risk Stratification and Individualized Patient Care in Uveal Melanoma. PLoS Curr. 2013 Apr 9;5:ecurrents.eogt.af8ba80fc776c8f1ce8f5dc485d4a618. doi: 10.1371/currents.eogt.af8ba80fc776c8f1ce8f5dc485d4a618. PMID: 23591547; PMCID: PMC3625622.
- Hiemcke-Jiwa LS, ten Dam-van Loon NH, Leguit RJ, et al. Potential Diagnosis of Vitreoretinal Lymphoma by Detection of MYD88 Mutation in Aqueous Humor With Ultrasensitive Droplet Digital Polymerase Chain Reaction. JAMA Ophthalmol. 2018;136(10):1098–1104.
- Nagarkatti-Gude N, Wang Y, Ali MJ, Honavar SG, et al. Genetics of primary intraocular tumors. Ocul Immunol Inflamm. 2012;20(4):244-254.
- Mehenaz Hanbazazh, Thaddeus P. Dryja (2020) Molecular Genetics of Intraocular Tumors, Seminars in Ophthalmology, 35:3, 174-181.
- Wen X, Lu L, He Z, Fan X. Orchestrating epigenetic roles targeting ocular tumors. Onco Targets Ther. 2016;9:1001-1009. Published 2016 Feb 29. doi:10.2147/OTT.S9399
