It is evident that GA potentially affects approximately 8 million people worldwide and is a rising cause of blindness in the developed world, specifically within the Caucasian population compared to other ethnicities.2,3
The "why" of geographic atrophy
The outstanding questions have been: how does this condition evolve, and where does it start? We begin by looking at the complement system implicated in GA via human genetics,
retinal pathology, genomic and proteomic analyses of human tissues, and work in numerous animal models.
4-12 The true identity lies within the details of complement activation in the disease—classical, lectin, and alternative pathways.
This article sets out to help you interactively understand the complement pathway through biochemical models, clinical time-lapse
fundus photography, fundus autofluorescence imaging studies, and finally, an appreciation of the visual impact of GA if left untreated.
The complement system
The complement cascade involves many protein interactions that occur within the plasma, on cell surfaces, and within cells.13 The cascade begins with three pathways: classical, lectin, and alternative. Each pathway converges on C3.14 Then C3 mediates multiple functions and eventually cleaves C5, which forms the terminal lytic pathway building up to the membrane attack complex (MAC).15,16
Of note, the alternative pathway (AP) is the oldest evolutionary signaling pathway of the three. The alternative complement pathway acts both independently of, and as an amplification loop for, the classical and lectin pathways.17 Shown in the diagram in regard to the AP amplification loop are key components of the C3 pathway, including C3 fragments, the complement receptors, and associated functions.
This portion of the illustration is meant to appreciate the myriad of different interactions between C3 fragments and their receptors on a generic cellular scaffold, which indirectly causes cell lysis leading to photoreceptor dropout, vision loss, and the formation of GA.14
The anaphylatoxins (C3a and C5a), their receptors, and associated functions activate and further heighten inflammation within the choriocapillaris/BM/RPE interface. The remainder of the Lytic pathway is played out in the final stage, as shown with these steps culminating with the MAC.14
The complement cascade
When RPE cells are exposed to healthy Bruch’s membrane (BM) and carry the CFH 402Y polymorphism in quiescent conditions, glucose is transported efficiently from the choroid/choriocapillaris (CC) through BM to the photoreceptors (PR) coupled with phagocyte photoreceptor outer segments (POS), and finally the elimination of oxidized lipids (OX) into the circulation. Integrins act as anchors between the RPE cells and BM, while FI and cofactors FH and FHL1 inhibit complement activation, and mitochondria respiration (OXPHOS = oxidative phosphorylation) is left intact.
AMD predisposition can be facilitated by the combination of genetic risk,
18 increasing age, and
external risk factors (i.e., smoking, obesity, etc.).
19,20 When RPE cells carrying the AMD-associated FH 402H polymorphism show mitochondrial dysfunction, while phagolysosomes (L) become edematous reflexively, causing “leaky” integrin interactions between the BM and RPE.
21,22 With these risk factors in mind, the BM extracellular matrix (ECM) illustrates modeling changes with apparent glucose transport impairment.
At this stage, complement turnover is stimulated by aberrant ECM and a lack of effective inhibition due to the presence of high-risk variants in FI, FH, FHL1, and the accumulation of FH antagonists, such as the FHR proteins.23,24 Complement turnover promulgates proinflammatory products to accumulate in the intercapillary septa and within BM itself.
Additionally, as mitochondrial oxidative stress boils over, it causes metabolic stress in the RPE cells, which can further stimulate inflammation.25 Hydrogen peroxide (H2O2) and products of lipid peroxidation engorge the already altered BM, promoting the upregulation of inflammatory cytokines and the high-risk CFH 402H variant with FHR accumulation amplifying this effect.26
Switching gears to early AMD, there is a continuous accumulation of oxidized lipids. Subsequently, complement activation products and MAC accumulation in drusen further promote an inflammatory response and immunological chain reaction. Localized inflammation, in conjunction with the release of anaphylatoxins C3a and C5a, causes the recruitment and activation of immune cells and mast cell degranulation into the choroid/BM/RPE interface.27
These inflammatory activities incrementally aggravate retina homeostasis, initiating RPE cell degeneration and the inability to support rod photoreceptor cells, which leads to retinal tissue damage and eventual morbidity.28
Complement activation from start to finish in geographic atrophy
When observing increased complement protein access to the outer neuroretinal space, it can be rooted back to the loss of RPE barrier function and damage to Bruch’s membrane.28,29 Further, reduced expression of membrane regulators of complement activation incites the complement attack on RPE cells and photoreceptors in the central foveal lesion and perifoveal border(s).
Subsequently, the generation of complement anaphylatoxins C3a, C4a, and C5a at the choroid/BM/RPE interface leads to C3 and C4 opsonization and formation of the MAC.14,27 Combining these complement activities together with an increased presence of phagocytic cells (macrophages and neutrophils) and mast cells at the interface promotes further loss of photoreceptors and RPE.27,30
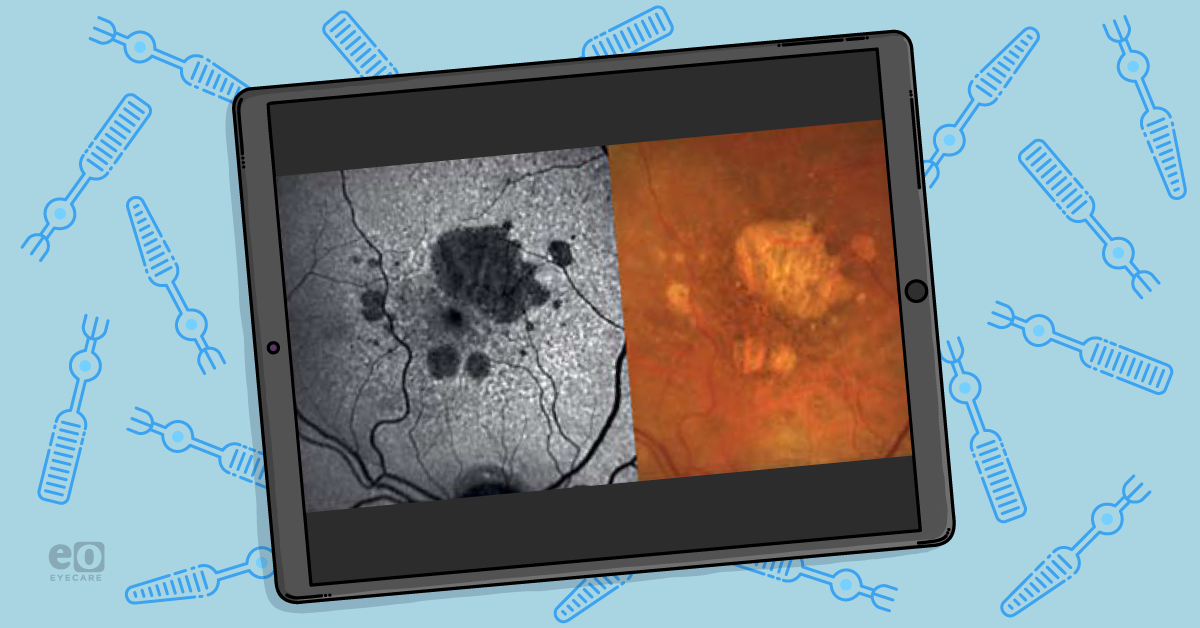
Here we see the development and progression of atrophy in purely nonexudative age-related macular degeneration with annual retinal imaging during 4-year follow-up from baseline.
The first row is a set of color fundus photographs (CFP), while the second row is confocal scanning laser ophthalmoscopy (CSLO) using blue-light fundus autofluorescence (FAF) for image capture.
FAF demonstrates areas of hyper-autofluorescence and hypo-autofluorescence. The photoreceptors shed damaged outer-segments, which are then ingested through phagocytosis by the RPE.
These molecules are stored in liposomes and form lipofuscin (LF). Areas of excess LF will demonstrate hyper-autofluorescence. This is also known as areas of “damaged” RPE cells.
However, upon RPE cell death or absence (atrophy), the LF disappears, and the tissue becomes hypofluorescent. Areas of GA with a hyperautofluorescence rim might be suggestive of faster GA lesion progression.31,32
As geographic atrophy is typically initiated outside of the foveal area, decreasing the rate of insulted GA area by 25% to 50% can potentially delay progression to the fovea by years.6 This is why early intervention is paramount for these patients to retain long-term vision.
Understanding the progression of GA
Flipping to an illustrative approach of GA area growth over time, the dotted circles in the image adjacent to the graph represent expected GA growth per expected natural history (red) or with reduced speed of progression (blue, green).
It is evident from the literature that the rate of geographic atrophy progression could be more rapid by approximately 2.8 times toward the periphery than toward the fovea.6,33 In addition, multifocal atrophic lesions progress at a faster rate than unifocal lesions. Larger geographic atrophy lesions grow quicker than smaller atrophic lesions.
GA lesions can significantly affect patient vision. Central vision is largely preserved until atrophy encroaches on the fovea, though tasks requiring full-field vision, such as reading/writing,
digital screens, driving, sewing, etc., can be impaired as peripheral vision is lost.
Of note, geographic atrophy often presents as functional vision loss before Snellen visual acuity will be lost.
Conclusion
As we begin to appreciate the nuances of the complement cascade, the identification of strategic
targets for GA therapy will continue to emerge from research centering around natural history and genetics.
Furthermore, it is crucial to expand the understanding of GA pathophysiology in order to understand the contributing factors that could lead to subsequent visual function decline. With increased knowledge and
novel therapeutics, we can potentially offer these GA patients hope for sight preservation.