




Corneal dystrophies are a rare and heterogeneous group of hereditary, non-inflammatory disorders caused by a range of deposited substances within the cornea. These dystrophies are typically delineated based on which anatomic location of the cornea is involved.
Briefly, the layers of the cornea, from the most superficial to deep, are as follows:
- Epithelium
- Bowman’s layer
- Stroma
- Descemet’s membrane
- Endothelium
Corneal dystrophies are further classified based on the level of evidence to support their respective existences as defined by The International Committee for Classification of Corneal Dystrophies.
Granular corneal dystrophy (GCD) is a stromal dystrophy driven by genetic variations in the TGFBI gene on the 5q31 locus.1 This results in the misfolding of the corresponding keratoepithelin protein, which is involved in the normal function of fibronectin, collagen, and integrins2 (found in corneal stromal tissue). The misfolded keratoepithelin subsequently deposits throughout the corneal stroma.
Given that the cornea provides two-thirds of the visual system’s refractive power3 and that the stroma accounts for approximately 90% of the corneal thickness,4 pathologies affecting the stroma may cause visually significant symptoms (inherited in an autosomal dominant manner).
GCD has two subtypes: GCD type I, or corneal dystrophy Groenouw type I; and GCD type II, or Granular-lattice dystrophy or Avellino corneal dystrophy. Different pathogenic variants cause these subtypes on the same gene locus described above.
Granular corneal dystrophy type I
Epidemiology
The precise incidence of GCD type I remains unknown; however one report from China estimated a local incidence of 0.24% in a cohort of 2068 refractive surgery candidates.5

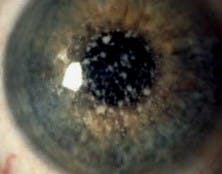
Figure 1: GCD type I (also corneal dystrophy Groenouw type I)
Signs and symptoms
Hallmark findings in GCD type I are typically identified on biomicroscopy. Discrete, irregular crumb- or flake-shaped white deposits within the central, anterior stroma are characteristic. These deposits are progressive, becoming increasingly numerous and coalescing over time, and may eventually result in impairments in visual acuity.6 Visual acuity is not usually affected until the 4th decade for most patients. Sparing of the limbus is expected. Corneal topography is initially unaffected, but patients may develop an uneven corneal surface with progression.7
Patients may report glare or photophobia symptoms; changes to visual acuity may occur later in life, usually in the 4th or 5th decade. Some may report recurrent corneal erosions due to involvement of the proximal corneal epithelial basement membrane.
Diagnosis and testing
The identification of protein deposits in the anterior stroma on biomicroscopy is classic. Anterior segment optical coherence tomography (OCT) may demonstrate hyperreflective anterior stromal deposits with unreflective shadows. A corneal biopsy may be pursued, and red hyaline material on Misson’s trichrome stain without Congo red stain. Confirmatory genetic testing shows variations in the TGFBI gene chromosome on 5q31. The most strongly associated genetic variation is p.Arg555Trp.9
Treatment and management
Conservative management is the mainstay therapy. Lubricating drops throughout the day and lubricating ointments, particularly at night, may reduce corneal erosions arising from the shear forces applied to the cornea upon eyelid opening after periods of overnight corneal drying. Bandage contact lenses may be considered for non-severe erosions. Tinted glasses or contacts are helpful in managing photophobia.
Surgical options
Surgical management begins with the least to increasingly more invasive options. Excimer ablation is an excellent candidate for managing superficial and often more visually significant corneal deposits.1,9 This does not affect future candidacy for keratoplasty and may even delay its necessity.9 When keratoplasty is required, deep anterior lamellar or penetrating keratoplasty are the surgical options of choice. In the case of recurrent disease, repeat keratoplasty is reasonable.
Prognosis
The stromal deposits characteristic in GCD type I are known to expand laterally and posteriorly over time slowly. A progressive course is typical. Post-keratectomy recurrence rates of corneal deposits are almost universal though reports indicate varying rates at which these deposits recur. Penetrating keratoplasty gives the longer freedom intervalm from significant losses to best-corrected visual acuity.10
Granular corneal dystrophy type II
Epidemiology
Like that of GCD type I, the precise incidence or prevalence of GCD type II is unknown due to its relative rarity. However, authors in South Korea have noted a prevalence of a common GCD type II genetic variation (p.R124H) as being 291.3 per 100,000.11 Similar estimates from the United States have not been reported. It is known as granular-lattice dystrophy. It displays findings of both diseases or Avellino dystrophy (first described in families from Avellino, Italy).

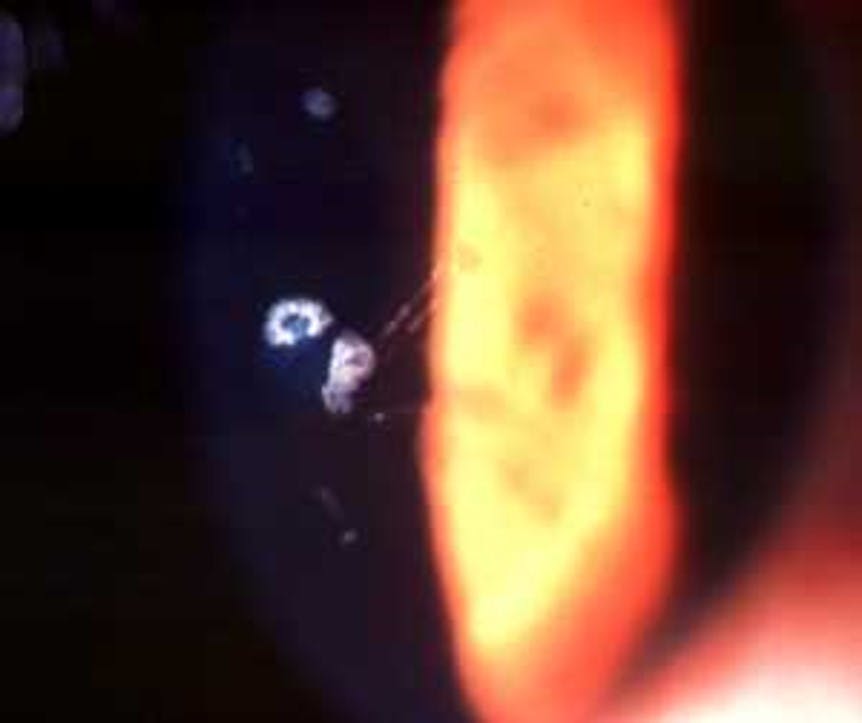
Figure 2: GCD type II (also Granular-lattice dystrophy or Avellino corneal dystrophy)
Signs and symptoms
GCD type II is characterized by both anterior stromal deposits and deeper stromal, amyloid lattice lesions.1 Other than these lattice lesions, many of the signs and symptoms of GCD type II not only mirror those of GCD type I but may be more familiar: photophobia, corneal erosions, and disturbances to visual acuity. However, corneal opacities and visual impairment typically manifest earlier, with patients noting vision loss in adolescence and requiring surgical management.9 Most patients maintain a vision of 20/70 or better, even late in the disease.
Diagnosis and testing
Protein deposits in the anterior stroma and lattice lesions in the deeper stroma are expected on biomicroscopy. Histopathology of biopsied corneal tissue shows red staining in the anterior stroma with Masson trichrome staining and may variably demonstrate Congo red staining in the deeper stroma suggestive of amyloid.1,12GCD type II also arises from genetic variations on chromosome 5q31, with the most strongly associated variation being p.Arg555Trp.9
Treatment and management
Aggressive lubrication is essential in reducing corneal erosions. Similar conservative therapeutic strategies for GCD type I aimed at protecting the ocular surface and reduce patient discomfort, such as bandage contact lenses and tinted glasses. Surgery is reasonable when patient symptoms are severe or a significant compromise to visual function.
Surgical options
Surgical management of GCD type II is similar to that of GCD type I. Phototherapeutic keratectomy (PTK) is useful in addressing anterior stromal opacities, but it typically starts from ablation margins and progresses centrally. When recurrence happens, penetrating keratoplasty may be considered when more diffuse and deeper corneal opacities exist; however, recurrences are still expected to occur. Laser in situ keratomileuses (LASIK) may help improve vision and delay corneal transplantation but has been noted to have a particularly strong association with causing mild exacerbations.13
Prognosis
Granular stromal deposits generally appear before lattice lesions and continue to worsen over time. These lattice lesions may coalesce to form linear-appearing opacities.14 Like GCD type I, recurrence is expected to occur within several years of surgical management, regardless of technique; however, in the case of PTK, recurrence may occur within a scale of months.15
Role of genetic testing
Genetic testing is a crucial component in diagnosing, understanding, and monitoring disease progression and establishing paths forward for future gene trials.
Genetic testing may be a useful confirmatory tool despite a diagnosis often being clear on examination, cases where phenotypic overlap exists between several closely related but distinct corneal dystrophies. Furthermore, a precise diagnosis may help patients and providers better understand and prepare for future disease progression and enable informed decision-making regarding treatment options and support services.
Lastly, in an era of precision medicine, understanding the spectrum of genetic variations associated with granular corneal dystrophy may pave a path toward standardizing future gene trials and eventual development of targeted gene therapy (as in the case of voretigene neparvovec-rzyl for biallelic RPE-65-associated Leber’s congenital amaurosis).
References
- Duker, and James J. Augsburger. Ophthalmology. Edinburgh: Mosby Elsevier, 2009
- Ivanov, S. V., Ivanova, A. V., Salnikow, K., Timofeeva, O., Subramaniam, M., Lerman, M. I. Two novel VHL targets, TGFBI (BIGH3) and its transactivator KLF10, are up-regulated in renal clear cell carcinoma and other tumors. Biochem. Biophys. Res. Commun. 370: 536-540, 2008.
- Sridhar MS. Anatomy of cornea and ocular surface. Indian J Ophthalmol. 2018;66(2):190-194. doi:10.4103/ijo.IJO_646_17
- Reinstein DZ, Archer TJ, Gobbe M, Silverman RH, Coleman DJ. Stromal thickness in the normal cornea: three-dimensional display with artemis very high-frequency digital ultrasound. J Refract Surg. 2009;25(9):776-786. doi:10.3928/1081597X-20090813-04
- Song Y, Sun M, Wang N, Zhou X, Zhao J, Wang Q, Chen S, Deng Y, Qiu L, Chen Y, Aldave AJ, Zhang F. Prevalence of transforming growth factor β-induced gene corneal dystrophies in Chinese refractive surgery candidates. J Cataract Refract Surg. 2017 Dec;43(12):1489-1494. doi: 10.1016/j.jcrs.2017.07.038. Epub 2017 Dec 9. PMID: 29233738.
- Lin ZN, Chen J, Cui HP. Characteristics of corneal dystrophies: a review from clinical, histological and genetic perspectives. Int J Ophthalmol. 2016;9(6):904-913. Published 2016 Jun 18. doi:10.18240/ijo.2016.06.20
- Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7. Published 2009 Feb 23. doi:10.1186/1750-1172-4-7
- Venkateswaran N, Galor A, Wang J, Karp CL. Optical coherence tomography for ocular surface and corneal diseases: a review. Eye Vis (Lond). 2018 Jun 12;5:13. doi: 10.1186/s40662-018-0107-0. PMID: 29942817; PMCID: PMC5996489.
- Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, Kim EK. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog Retin Eye Res. 2016 Jan;50:67-88. doi: 10.1016/j.preteyeres.2015.11.002. Epub 2015 Nov 28. PMID: 26612778.
- Lewis DR, Price MO, Feng MT, Price FW Jr. Recurrence of Granular Corneal Dystrophy Type 1 After Phototherapeutic Keratectomy, Lamellar Keratoplasty, and Penetrating Keratoplasty in a Single Population. Cornea. 2017 Oct;36(10):1227-1232. doi: 10.1097/ICO.0000000000001303. PMID: 28749898.
- Park JE, Yun SA, Roh EY, Yoon JH, Shin S, Ki CS. Prevalence of granular corneal dystrophy type 2-related TGFBI p.R124H variant in a South Korean population. Mol Vis. 2021 May 8;27:283-287. PMID: 34012230; PMCID: PMC8116257.
- Kim TI, Roh MI, Grossniklaus HE, Kang SJ, Hamilton SM, Schorderet DF, Lee WB, Kim EK. Deposits of transforming growth factor-beta-induced protein in granular corneal dystrophy type II after LASIK. Cornea. 2008 Jan;27(1):28-32. doi: 10.1097/ICO.0b013e318156d36d. PMID: 18245963.
- Jun I, Jung JW, Choi YJ, Kim TI, Seo KY, Kim EK. Long-term Clinical Outcomes of Phototherapeutic Keratectomy in Corneas With Granular Corneal Dystrophy Type 2 Exacerbated After LASIK. J Refract Surg. 2018 Feb 1;34(2):132-139. doi: 10.3928/1081597X-20171220-01. PMID: 29425392.]
- Holland EJ, Daya SM, Stone EM, Folberg R, Dobler AA, Cameron JD, Doughman DJ. Avellino corneal dystrophy. Clinical manifestations and natural history. Ophthalmology. 1992 Oct;99(10):1564-8. doi: 10.1016/s0161-6420(92)31766-x. PMID: 1454323.
- Moon JW, Kim SW, Kim TI, Cristol SM, Chung ES, Kim EK. Homozygous granular corneal dystrophy type II (Avellino corneal dystrophy): natural history and progression after treatment. Cornea. 2007 Oct;26(9):1095-100. doi: 10.1097/ICO.0b013e3181484013. PMID: 17893542.

