


Patient background
A 60-year-old female was referred for the evaluation of pigmented retinal lesions and a glaucoma workup. The referring optometrist had noted bilateral moderate cupping and numerous small to medium lesions of varying pigmentation in both eyes. The patient denies ever being treated for glaucoma or being told that she has high IOP. Likewise, she denied any past signs/symptoms consistent with intraocular inflammation.
Her best-corrected vision was mildly reduced to 20/25- OD, OS due to mild nuclear sclerosis and anterior cortical spoking. IOP was consistently under 18mmHg on every visit. Central corneal thickness with ultrasound pachymetry measured 552μm and 549μm. Angles were open to scleral spur 360 degrees OD, OS without any abnormalities.
A dilated fundus examination revealed 0.60 cupping with possible rim thinning inferiorly in the right eye and 0.55 cupping in the left. No pallor or RNFL defects were appreciated. In the right eye, two small pigmented lesions with a depigmented halo were noted temporal to the fovea. A similar but larger lesion was noted superior-temporal to the posterior pole. Pinpoint pigmented lesions were scattered throughout the periphery as well. Multiple similar lesions were noted in the left posterior pole and midperipheral retina (as seen in Figures 1 and 2).
Numerous less distinct and lightly pigmented lesions were noted superior to the posterior pole in the left eye. Apart from the sectoral grouping of the less distinct and lightly pigmented lesions in the left eye, no particular distribution pattern of the darkly pigmented lesions was noted. All the lesions were flat without orange pigmentation fluid, overlying vitreal abnormalities, or associated inflammation.
Figures 1 and 2 demonstrate RPE hamartomas associated with familial adenomatous polyposis (RPEH-FAP) of varying sizes and pigmentation noted in both eyes. Note the irregular depigmented regions often referred to as a fish-tail or comet configuration.


Figure 1, image courtesy of Daniel Epshtein, OD, FAOO.

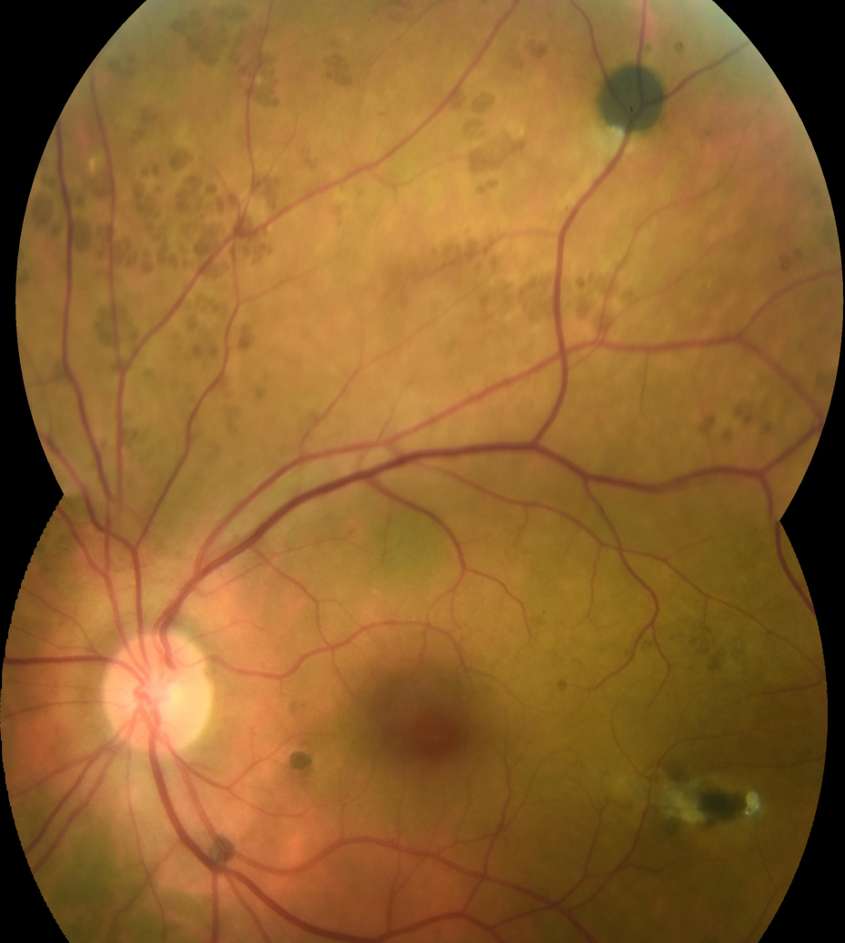
Figure 2, image courtesy of Daniel Epshtein, OD, FAOO.
OCT imaging of the various darkly pigmented lesions revealed a predominantly normal RPE, though some lesions exhibited pigment migration and hypertrophy. All the darkly pigmented lesions had varying degrees of overlying ellipsoid zone and outer retinal degeneration (as seen in Figure 2).
The lesions with the greatest amount of outer retinal degeneration also appeared to have small pockets of fluid reminiscent of outer retinal cavitations noted in macular telangiectasis type 2, cone dystrophy, and achromatopsia. The less distinct and lightly pigmented lesions noted superior to the posterior pole in the left eye were not visualized with OCT imaging.
Figures 3 and 4 show OCT imaging revealing variable RPE and outer-retinal degeneration through small and medium RPEH-FAP.

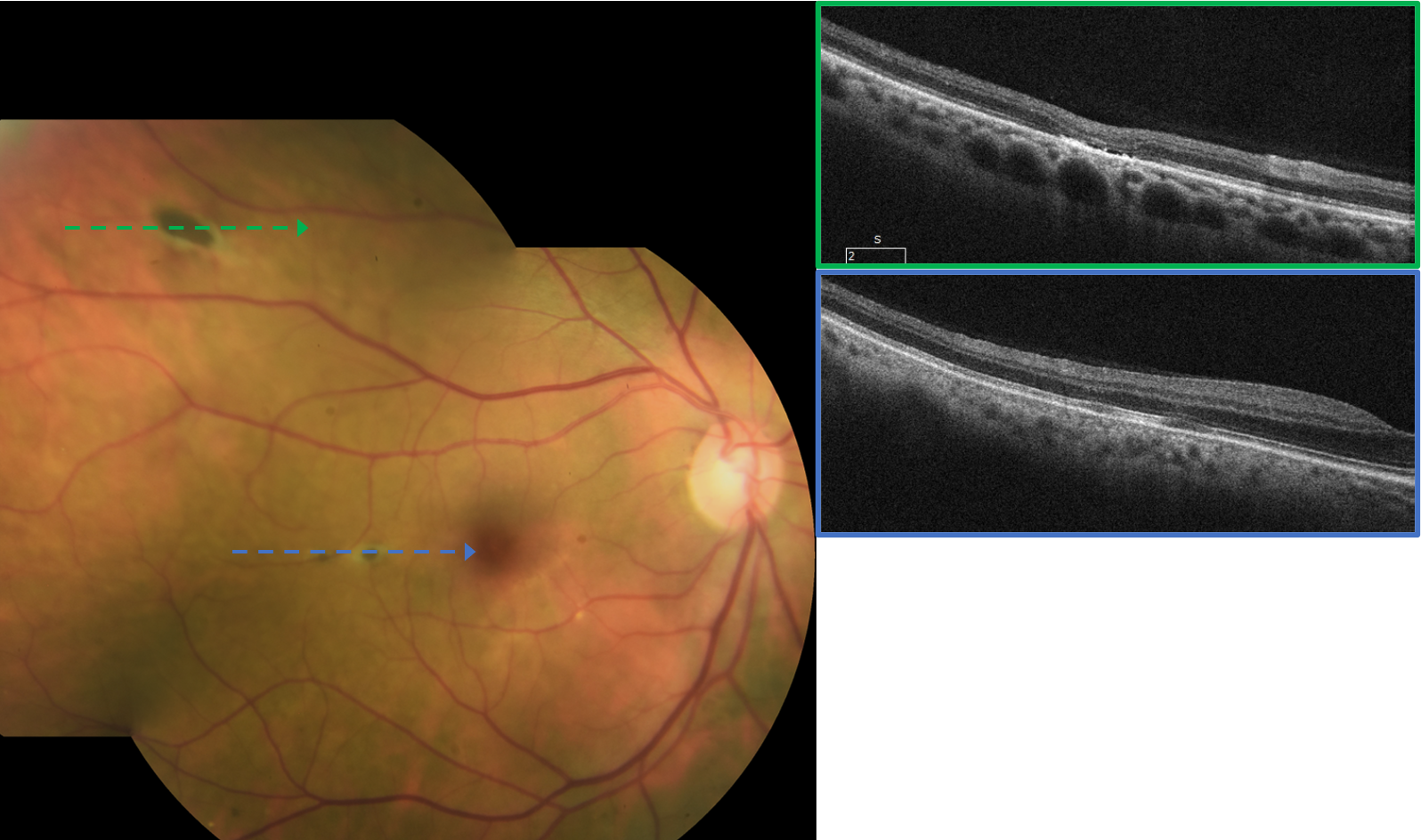
Figure 3, image courtesy of Daniel Epshtein, OD, FAOO.


Figure 4, image courtesy of Daniel Epshtein, OD, FAOO.
Though these retinal lesions looked unusual, a quick review of the patient’s medical history immediately revealed the diagnosis. The patient was diagnosed with colon cancer 26 years prior. As part of the investigation for her colon cancer, she was diagnosed with familial adenomatous polyposis (FAP) and eventually with Gardner’s syndrome due to the presence of characteristic benign skin tumors and osteomas.
She had undergone partial colon resection along with chemotherapy. Luckily, she was currently in remission but required close monitoring with frequent endoscopies and colonoscopies. Her father, paternal grandmother, and two brothers were all diagnosed with colon cancer and FAP as well.
Familial adenomatous polyposis
Familiar adenomatous polyposis (FAP) is an autosomal dominant inherited disorder primarily characterized by numerous colorectal polyps.1 FAP is caused by mutations in the adenomatous polyposis coli (APC) gene, which codes for tumor suppressor genes.1 Without a properly functioning APC gene, FAP patients develop hundreds to thousands of precancerous colorectal polyps that eventually progress to colorectal carcinoma in almost 100% of patients.1
By age 15, 50% of FAP patients develop colorectal polyps, increasing to 95% by age 35.1 Without treatment, patients with FAP tend to develop colorectal carcinoma by age 35 to 40.1 Up to 70% of patients have a family history of FAP, while 20 to 30% of cases develop due to a de novo mutation.1
Though most notably known for associations with colorectal cancer and other gastrointestinal findings, FAP patients are also at higher risk for extracolonic findings, such as brain tumors, thyroid carcinomas, cutaneous growths, and osteomas.
Familial adenomatous polyposis versus Gardner’s syndrome
Gardner’s syndrome is a phenotypic subtype of FAP in which patients develop osteomas, epidermoid cysts, fibromas, and desmoid tumors in addition to the classic FAP colorectal findings. Gardner’s syndrome is a clinical syndrome and is not diagnosed based on genetic testing. For this reason, the term Gardner’s syndrome has fallen out of favor with some clinicians and scientists.
Retinal lesions in FAP
Though often referred to as congenital hypertrophy of the retinal pigment epithelium (CHRPE), multifocal CHRPE, or bear tracks, the retinal lesions associated with FAP are distinct lesions not associated with CHRPE or any of its variants.2 The current nomenclature for the retinal lesions noted in FAP is RPE hamartomas associated with familial adenomatous polyposis (RPEH-FAP).2 Another less specific term, pigmented ocular fundus lesions (POFL), can also be found in the literature.
RPEH-FAP are noted in 70% of FAP patients and are most often multifocal, bilateral, and without any specific distribution pattern.2 The individual lesions vary in appearance but are typically ovoid with light to dark pigmentation and an irregularly depigmented tail. The lesions range in size from pinpoint to several disc diameters in length.
It is thought that RPEH-FAP are benign, nonprogressive lesions present at birth.1,2 Periodic ocular monitoring is recommended, but any affected patient should be scrutinized for colon cancer and/or APC mutations.
Back to the patient
After reviewing the patient’s medical history and confirming that several specialists closely monitor her, she was assured that the retinal lesions are benign and do not require any intervention. A complete glaucoma workup, including pachymetry, perimetry, OCT imaging, and gonioscopy, was performed. All testing was unremarkable, and the patient was deemed a low-risk glaucoma suspect and educated to follow up yearly for repeat glaucoma testing and evaluation of RPEH-FAP.
Though no specific guidelines exist, RPEH-FAP lesions are thought to be relatively stable and should be evaluated with funduscopy no less than once a year. Ancillary testing such as fundus autofluorescence and OCT can help diagnose RPEH-FAP, but ocular imaging does not need to be repeated unless clinical changes are noted.
Final thoughts
Familial adenomatous polyposis and its variants, such as Gardner’s syndrome, are conditions that all eyecare providers learn about but rarely see in practice. It is important to stay vigilant when evaluating chorioretinal lesions because a zebra like FAP may end up in your chair, and you may end up coordinating a referral cascade that eventually saves the patient’s life.
