



Central serous chorioretinopathy is an old condition getting a lot of attention recently. As our understanding of central serous chorioretinopathy evolved, so did our terminology. Originally described in the 19th century as a recurrent central retinitis, then capillarospastic central retinitis, central angiospastic retinopathy and central serous retinopathy. After careful examination of fluorescein angiographic data, Dr. Gass popularized the term central serous chorioretinopathy to represent the condition’s choroidal etiology.
Though Gass’s terminology has stuck around, the diagnostic criteria, nomenclature, and pathophysiological theories have advanced greatly in the past few decades.
Diagnosis and classification
Up until recently, there were no standardized diagnostic criteria for the diagnosis of central serous chorioretinopathy. All clinicians knew what central serous chorioretinopathy was, but we couldn’t point to a single widely accepted consensus paper to guide our diagnosis. In 2020, the Central Serous Chorioretinopathy International Group created specific diagnostic criteria (see below) to accurately diagnose central serous chorioretinopathy. Two major criteria, along with one of three minor criteria must be fulfilled to diagnose central serous chorioretinopathy.
Major criteria (must fulfill both)
- Presence or evidence of prior serous retinal detachment documented on OCT involving the posterior pole unrelated to other disease
- At least one area of RPE alteration on fundus autofluorescence, OCT, or infrared imaging
Minor criteria (must fulfill at least 1)
- Mid-phase hyperfluorescence placoid areas on indocyanine green angiography
- One or more focal leaks on fluorescein angiography
- Subfoveal choroidal thickness ≥400µm (consider age and axial length)
Figure 1 illustrates simple, primary central serous chorioretinopathy with foveal involvement in the right eye. Note a small area of pachychoroid pigment epitheliopathy in the left eye.

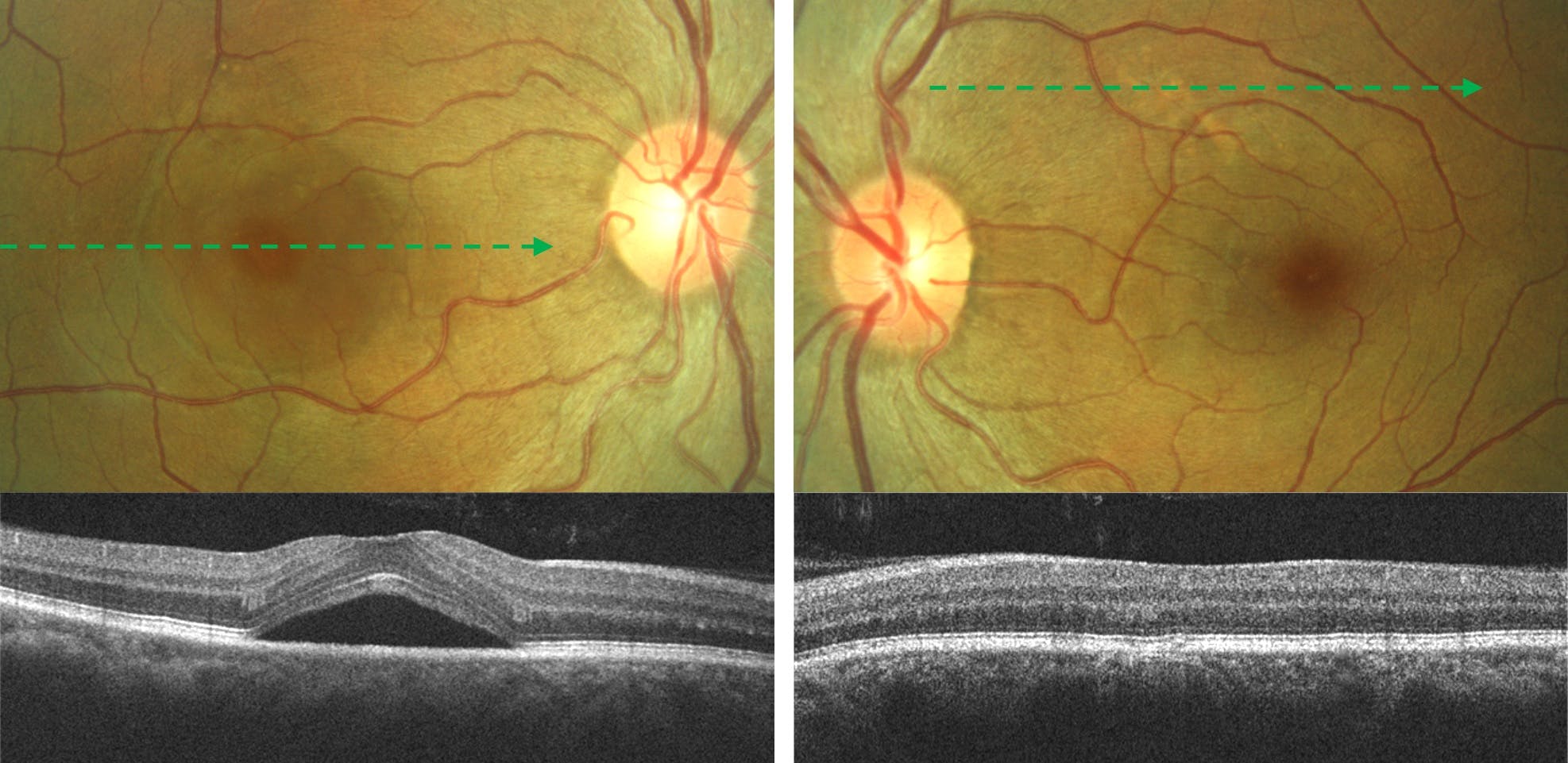
Figure 1
The self-limiting unilateral macular serous retinal detachment is the most common form of central serous chorioretinopathy, but a wide variability of presentations exist. The various subtypes of central serous chorioretinopathy based on the chronicity/extent of subretinal fluid and presence of retinal/RPE changes. Along with the above diagnostic criteria, the Central Serous Chorioretinopathy International Group created a classification system for the various forms of this disease.
Though daunting at first glance, the table below summarizes the classification of central serous chorioretinopathy subtypes and allows for reproducible and consistent classification.

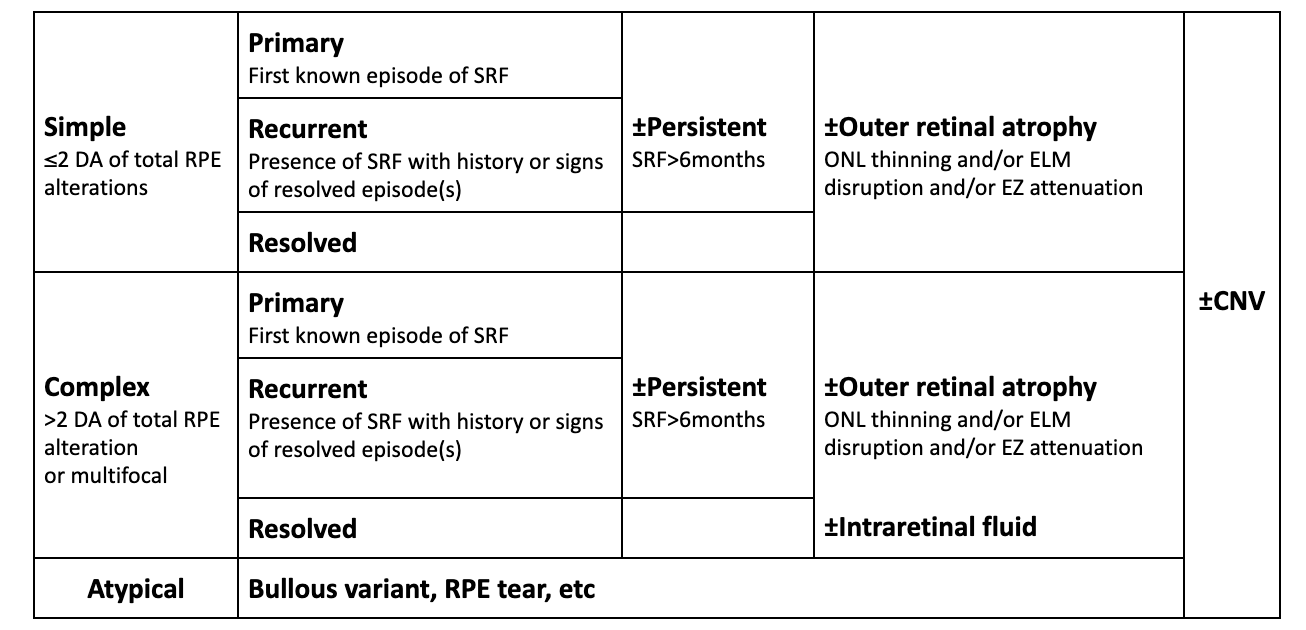
+ if fovea is involved (SRF, outer retinal atrophy, PED)
Following this algorithmic approach, a multimodal imaging approach must be utilized to properly classify central serous chorioretinopathy. OCT is the gold standard for detecting subretinal fluid and retinal atrophy. OCT angiography or dye-based angiography can be used to evaluate for the presence of choroidal neovascularization. Fundus autofluorescence, particularly ultra-widefield fundus autofluorescence, is a non-invasive modality for evaluating RPE alterations.
Figure 2 demonstrates complex, resolved central serous chorioretinopathy with outer retinal atrophy.

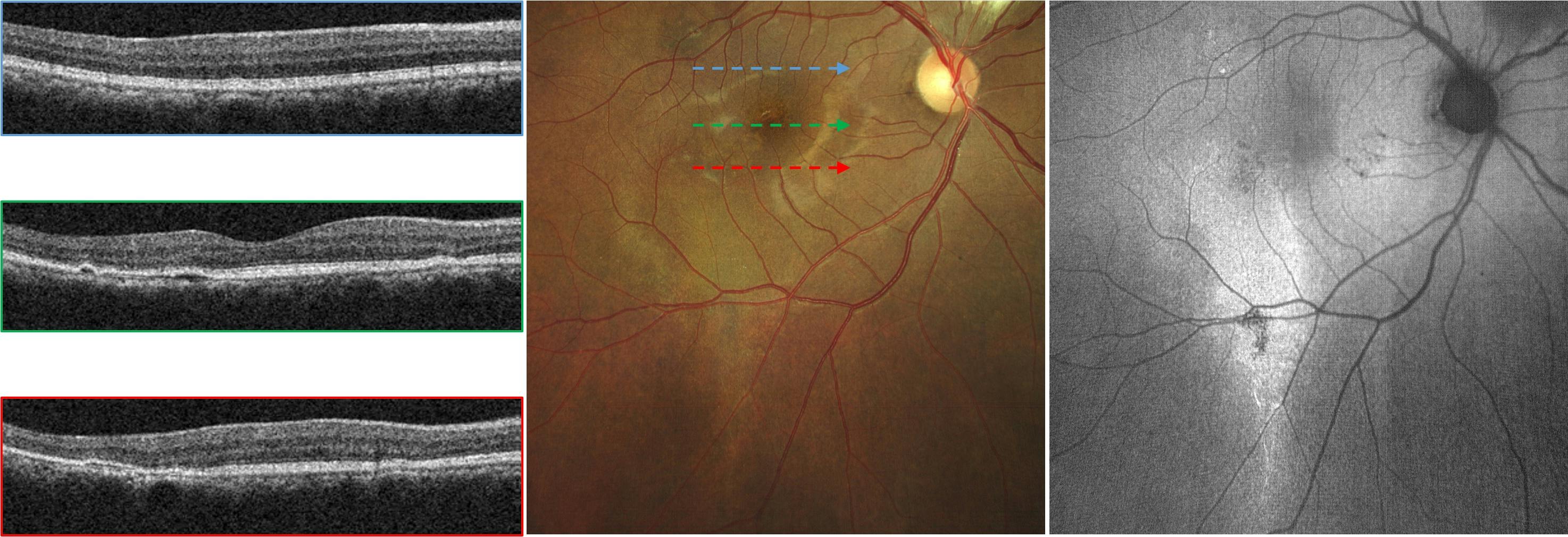
Figure 2
Etiology and pachychoroid
The etiology of central serous chorioretinopathy is not well defined but choroidal dysfunction is thought to play a key role in its pathogenesis. The associations of exogenous and endogenous hypercortisolism, stress, and Type A personalities with central serous chorioretinopathy have been known for many years but direct pathophysiological links have still not been ascertained.
Recent multimodal investigations have emphasized choroidal hyperpermeability with subsequent retinal pigment epithelium decompensation (whether physical or biochemical) and exudation into the subretinal space as the causative factor in central serous chorioretinopathy.
A new theory has suggested that choroidal hyperpermeability is caused by dysfunction at the level of the vortex veins. Changes in vortex vein anatomy, including anastomoses and dilatations, lead to the cascade of chorioretinal changes ending in central serous chorioretinopathy. The choroidal and vortex vein changes have also been found in other similar chorioretinopathies, giving credence to the theory that central serous chorioretinopathy belongs to the pachychoroid disease spectrum.
This family of diseases includes pachychoroid pigment epitheliopathy, pachychoroid neovasculopathy, and pachychoroid aneurysmal Type 1 neovascularization (previously known as polypoidal choroidal vasculopathy). The pachychoroid disease spectrum is characterized by diffuse or focal thickening of the choroid, choroidal hyperpermeability, dilated vessels within Haller’s layer of the choroid, inner choroidal atrophy, and reduced fundus tessellation.
Central serous chorioretinopathy
- Serous retinal detachment +/- pigment epithelial detachment +/- neovascularization
Pachychoroid pigment epitheliopathy
- RPE alterations +/- pigment epithelial detachments colocalizing to pachyvessels
- No serous retinal detachment
Pachychoroid neovasculopathy
- Irregular shallow pigment epithelial detachments secondary to sub-RPE/Type 1 neovascularization
Aneurysmal pachychoroid neovascularization (polypoidal neovasculopathy)
- Pachychoroid neovasculopathy that progresses to form aneurysmal structures
Focal choroidal excavation
- Choroidal excavation without posterior staphyloma or scleral ectasia
Peripapillary pachychoroid syndrome
- Choroidal thickening in the peripapillary area as opposed to the macular area
- May lead to peripapillary intraretinal fluid and/or disc edema.
Figure 3 represents pachychoroid neovasculopathy. Note the shallow irregular pigment epithelial detachment corresponding to the choroidal neovascularization on OCTA.

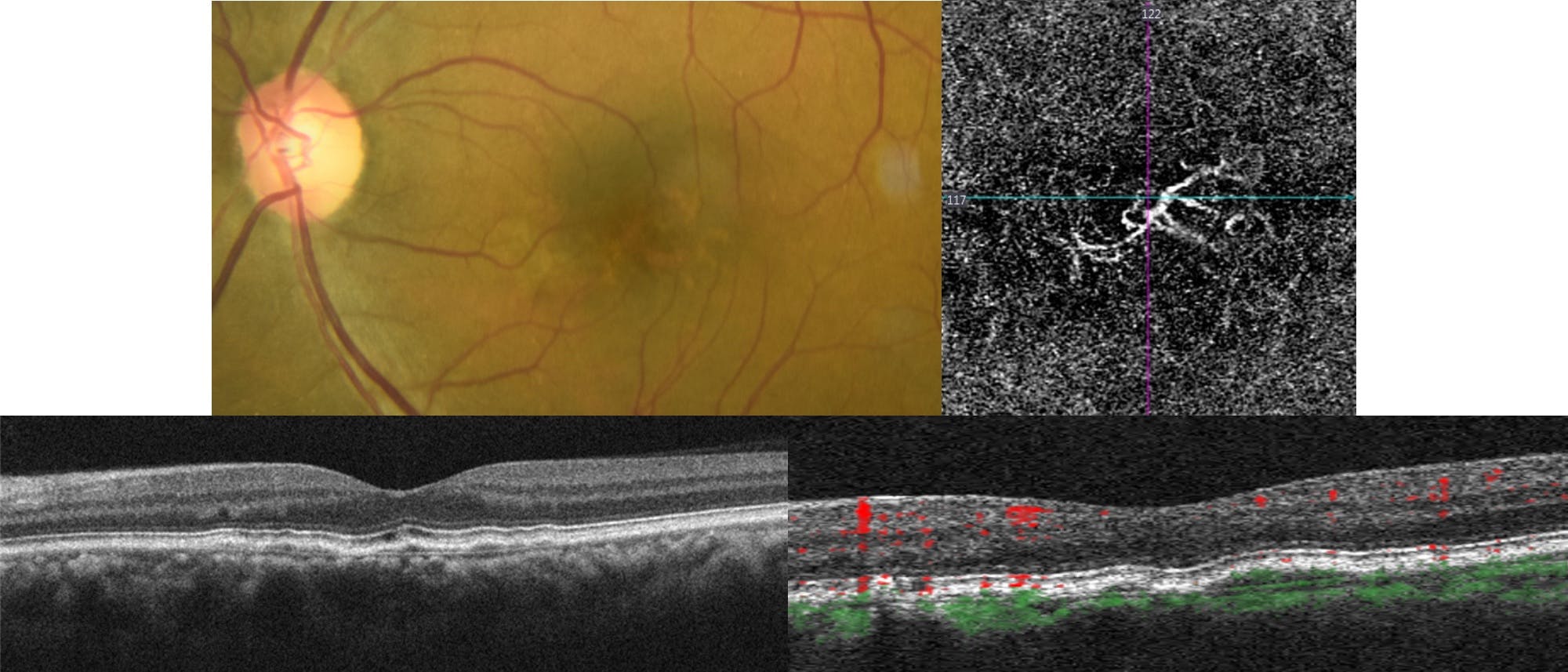
Figure 3
Central serous chorioretinopathy management
Though there is no standardized approach to central serous chorioretinopathy management and treatment, some clinical tendencies do exist. Spontaneous resolution is noted within several months in a majority of cases of acute central serous chorioretinopathy. For this reason, even if there is suspicion that the current presentation may be a reoccurrence or a foveal presentation of previously extramacular disease, observation is usually the first choice in management.
In cases of exogenous hypercortisolism, discussion with the patient’s prescribing physician should be initiated to discontinue or taper glucocorticoid treatment. It is important to rule out secondary choroidal neovascularization in these cases as that may require more rapid treatment.
In cases of persistent, recurrent, or chronic serous detachment, angiographic investigation can help guide treatment. Depending on symptoms, signs, chronicity of fluid, and the “type” of central serous chorioretinopathy various treatment protocols including laser photocoagulation, photodynamic therapy, and mineralocorticoid receptor antagonists have been used with varying degrees of success.
In Figure 4, we see focal choroidal excavation in a patient with pachychoroid neovasculopathy.

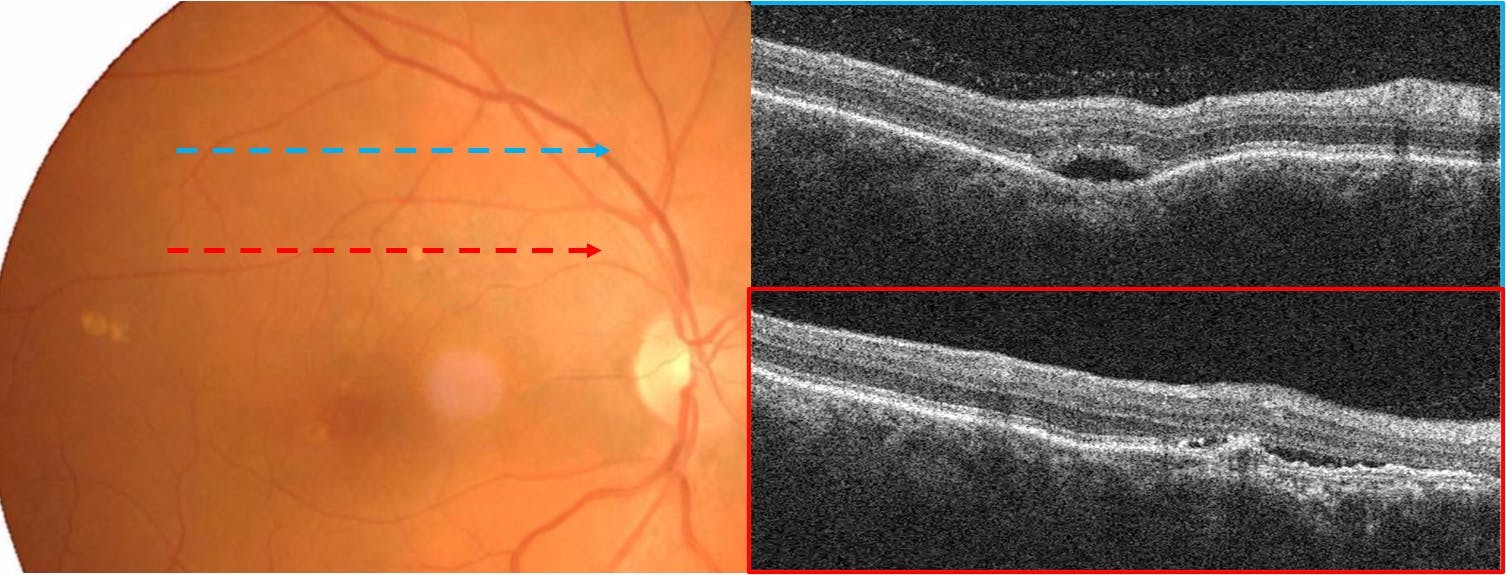
Figure 4
Though there is much that we still do not understand about central serous chorioretinopathy, the standardization of its diagnosis and classification represents a major step forward in improving our management of the condition. Combining the latest research with new innovative diagnostic modalities, clinicians can further optimize their treatment protocols and add to the ever-evolving literature of central serous chorioretinopathy.
