





With the aim of spreading awareness about rare diseases and promoting greater access to care, Rare Disease Day is observed each year on the last day of February. In the United States, a disease is considered rare if it affects less than 200,000 Americans.1
On average, ophthalmologists see certain conditions, such as glaucoma, cataracts, dry eye disease, and age-related macular degeneration, on a regular basis. However, as in all medical fields, doctors are inevitably confronted with much less common diseases that require innovative diagnostic and treatment approaches.
We asked three top ophthalmologists to weigh in on the rarest diseases they have encountered.
Timothy Murray, MD, MBA, discusses intraocular medulloepithelioma
Usually occurring in children between the ages of 2 and 10, intraocular medulloepithelioma is a nonhereditary neoplasm that frequently involves the ciliary body and appears as a greyish-white lesion with intratumoral cysts.2 Pediatric patients initially experience vision loss, leukocoria, conjunctival congestion, and rarely pain.2
Just how rare is intraocular medulloepithelioma?
Dr. Murray: The tumor can typically resemble retinoblastoma, which is the most common primary eye cancer in children. Even with that, there's only 300 cases of retinoblastoma in the United States a year; but for every 100 cases of retinoblastoma, there's less than one case of medulloepithelioma, so most doctors can practice an entire career and never see a case of this disease.
How does the condition present?
Dr. Murray: The problem is that these patients are often pre-verbal, so they're not capable of telling their parents that something is wrong. Children, typically, even when they're old enough to express a problem, don't always appreciate that they've lost vision in one eye because the other eye steps up.
The most common finding here is leukocoria, which is a white pupil. Normally, when you shine a light in the dilated pupil, it reflects back red. In leukocoria, the reflection comes back dull and typically white. The tumor usually starts in the ciliary body, which is anterior, and has fimbriated extension, making it appear as if little pseudopods are growing out of the tumor. That's very different from retinoblastoma.
Which exam techniques and tests were used for diagnosis?
Dr. Murray: We perform both a clinical exam and imaging on the child. To evaluate kids at this age, who will not allow you to pull and prod, we usually have to go to the operating room and perform an examination under anesthesia, so you're weighing the risk against the benefits.
The most important imaging for medulloepithelioma is ultrasound. On ultrasound, you often see echo-lucencies that look like a cyst not typically seen in retinoblastoma. Even saying that, people will mistake the diagnosis, at times, and manage the child incorrectly because of how rare this is.
What is the treatment protocol for intraocular medulloepithelioma?
Dr. Murray: We observe—we watch to make sure the child doesn't develop glaucoma, a retinal detachment, neovascular changes, abnormal blood vessels with leakage, or bleeding.
The most important aspect is ongoing imaging. We look at the eyes with ultrasound, which lets us look behind and around the wall. I see these patients back after 2 months, then 3 months, and then every 4 months, typically for 1 to 2 years. Next, we move to an every 6-month schedule—but I watch these kids forever.
We also do MRI scans on a regular basis to make sure we're not missing anything during our exam. Typically, we'll do an MRI at 3 months, then 6 months, then every 6 months, and then once a year. One of the other important things is to put them in protective eyewear; they wear glasses all the time, because they have one good eye and one eye with medulla epithelioma.
What has been your most interesting case report of intraocular medulloepithelioma?
Dr. Murray: I saw a baby that was referred for retinoblastoma at 1.5 years old. When we took a closer look, we found all of the clinical and imaging features for medulloepithelioma, so I informed the parents, “This is a medulloepithelioma, and the right thing to do is watch.” And that is what we did.
Figures 1 and 2: Imaging of the pediatric patient revealing intraocular medulloepithelioma.


Figure 1: Courtesy of Timothy Murray, MD, MBA.


Figure 2: Courtesy of Timothy Murray, MD, MBA.
That child is no longer a child; she is now 23 years of age. She saw us 6 months ago, and the vision in that eye had dropped to being legally blind because the tumor had caused a cataract. If there's long-term stability of these eyes, you can consider surgery.
So we had a discussion about whether we should operate or not. I told her there was not a right or wrong answer; however, she's a beautiful young lady and didn't like the appearance of the cataract and couldn't see. So, we operated on her 3 months ago, and she is now 20/20 in the eye with the medulloepithelioma, which shows that if you're cautious early and observe, you have options to manage later.
Figure 3: Intra-operative image of cataract surgery.


Figure 3: Courtesy of Timothy Murray, MD, MBA.
Figure 4: Post-operative results of cataract surgery.


Figure 4: Courtesy of Timothy Murray, MD, MBA.
In the past, her eye would have been removed; doctors nearly always opted for enucleation. We now know we don't need to do that unless there's a concern that the tumor is permanently damaging the eye, causing the eye to become painful, or is going to end up out of the eye—none of which was the case for her.
Do you have any clinical pearls for other ophthalmologists confronted with this condition?
Dr. Murray: Opting to biopsy the tumor (or repair the cataract) and leave the eye in place can result in the tumor growing out of the eye—that's when these children can die. If you think it's a malignant tumor, send it to a specialist who has better imaging ability or better clinical experience, because you don't want to make that mistake.
The problem is not being uncertain; the problem is being uncertain and not taking the next step, which is having the child imaged appropriately or sending them to an ocular oncologist. In my mind, you never get in trouble with a referral, because now you've shared the burden—and if you refer to somebody more expert and there's a problem, it's now the expert’s problem, not yours.
If it worries you enough to refer, even if it ends up being something trivial, you still did the right thing.
Anat Galor, MD, discusses Schnyder’s corneal dystrophy
This rare corneal dystrophy was first known as Schnyder crystalline corneal dystrophy, but after it was realized that only 50% of affected patients have corneal crystals, the name was changed by the International Committee for Classification of Corneal Dystrophies.3
Characterized initially by central corneal haze and/or crystals, Schnyder corneal dystrophy (SCD) also involves the subsequent appearance of arcus lipoides in the development of midperipheral haze.3 SCD leads to progressive vision loss.
Figure 5: Recurrent SCD; notice the arcus in the periphery.


Figure 5: Courtesy of Anat Galor, MD.
How does the condition present?
Dr. Galor: Corneal haze that presents around age 40 and progresses with time. A dense arcus peripherally accompanies the central haze. Some but not all individuals have corneal crystals within the haze.
Figure 6: A patient with Schnyder’s corneal dystrophy prior to any surgical intervention.


Figure 6: Courtesy of Anat Galor, MD.
Which exam techniques and tests were used for diagnosis?
Dr. Galor: This diagnosis can be made by slit lamp examination. However, a genetic test can confirm the diagnosis but is not needed.
What treatment options did you utilize?
Dr. Galor: Unfortunately, we do not have disease-modifying therapies for Schnyder's corneal dystrophy at this time. As such, we use various corneal procedures to restore corneal clarity when the corneal haze reaches a level where it significantly impacts function.
This oftentimes involves a deep anterior lamellar keratoplasty (DALK) procedure, given the full-thickness nature of the deposits. In addition, because Schyders is a progressive genetic disease, the deposits slowly return after surgery, and repeat procedures are often needed within a person’s lifetime.
Do you have any clinical pearls for other ophthalmologists confronted with this condition?
Dr. Galor: This is a rare presentation, but the diagnosis can be made by recognizing salient features at the slit lamp. Examining family members is often helpful, as this is an autosomal dominant disease.
Leonard Seibold, MD, discusses iris melanoma
Accounting for only 3 to 10% of uveal melanomas, the iris is the rarest primary site for a malignant melanoma.4 These uveal malignancies originate from melanocytes in uveal tissue and are usually composed of spindle cells.
The majority of iris melanomas—approximately 75%—originate in the inferior half of the iris. It is believed a primary risk factor for the development of iris melanoma is UV exposure in light-colored irides.5
How did the condition present?
Dr. Seibold: The condition presented as anterior uveitis and secondary open-angle glaucoma with unilateral significant elevation of intraocular pressure (IOP) to the mid-40s, significant anterior chamber pigment, and increased pigmentation of the iris.
Figure 7: Slit-lamp photographs of study patients showing different morphological features of iris melanoma.
The descriptions of the images below are as follows:
- A: Flat, weakly pigmented iris melanoma with a small ectropion uveae
- B: Dome-shaped, markedly pigmented iris melanoma with an irregular surface
- C: Nodular, moderately pigmented iris melanoma
- D: Ring-shaped, markedly pigmented iris melanoma visible temporally and inferiorly
- E: Diffuse iris melanoma with multiple pigmented areas
- F: Amelanotic, vascularised iris melanoma
- G: Tapioca iris melanoma with multiple small, amelanotic nodules on the surface
- H: Large iris melanoma leading to pupillary distortion and ectropion uveae

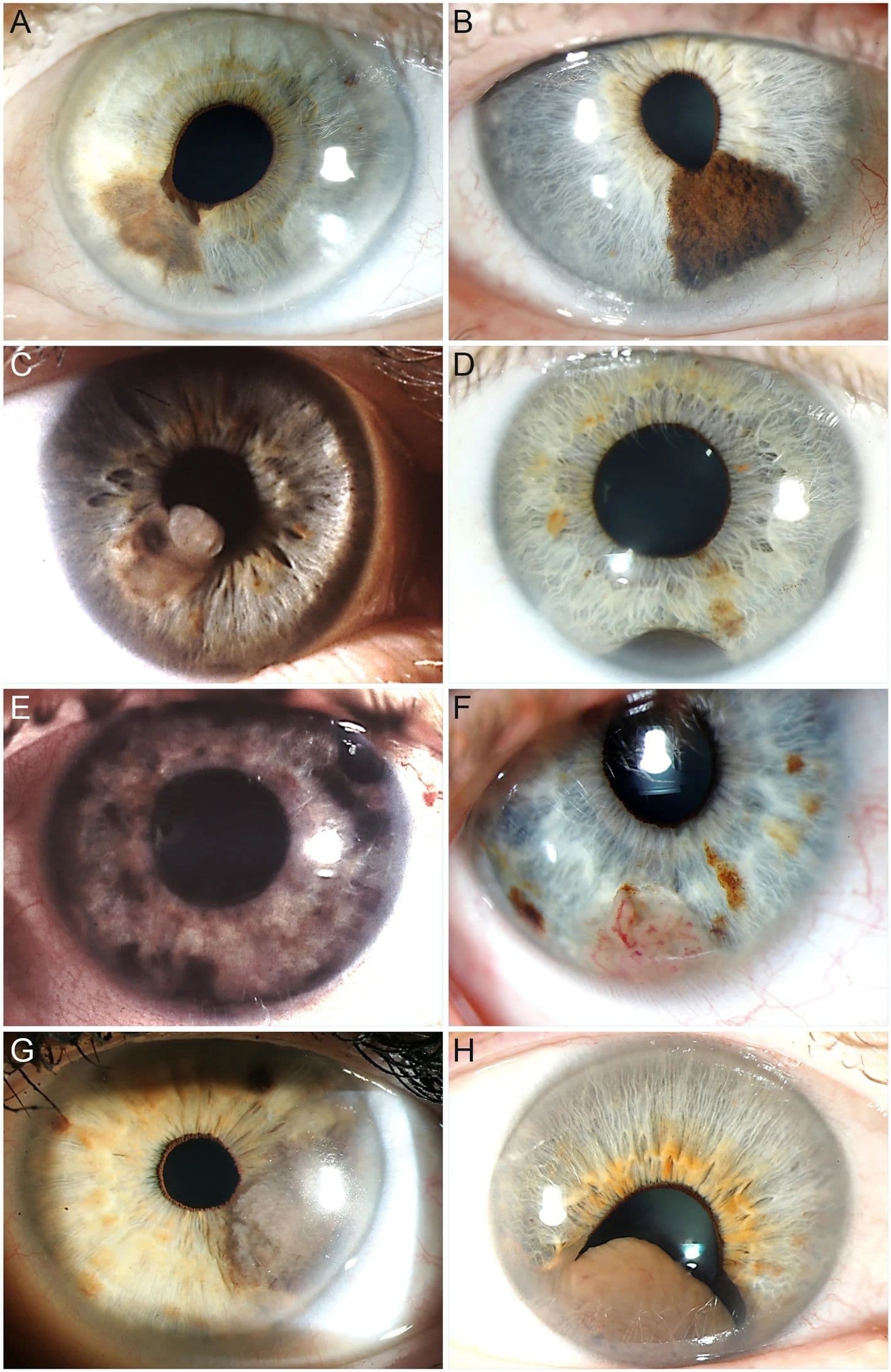
Figure 7: Morphological features of iris melanoma© Jørgen Krohn, Kristoffer Våge Sundal, Torbjørn Frøystein. Used under CC BY 4.0.
Which exam techniques and tests were used for diagnosis?
Dr. Seibold: The slit lamp examination was used to identify increased iris pigmentation, significant anterior chamber pigmented cells, and corneal edema from the elevated IOP. Gonioscopy identified heavy diffuse pigmentation of the angle without synechiae.
Ultrasound biomicroscopy was performed but failed to identify significant iris or ciliary body changes. Ultimately, anterior chamber paracentesis and iris biopsy were performed in the operating room to confirm the diagnosis of melanoma.
What treatment options did you utilize?
Dr. Seibold: Since the melanoma was diffusely infiltrated in the anterior chamber along with severe secondary glaucoma, the patient elected for enucleation of the eye, providing a definitive cure.
Do you have any clinical pearls for other ophthalmologists confronted with this condition?
Dr. Seibold: This patient was misdiagnosed with iritis and pigment dispersion syndrome by multiple physicians. Maintain a high suspicion for malignancy when pigment dispersion is unilateral and there are no other signs of inflammation or readily identifiable causes.
In conclusion
With the latest diagnostic techniques, imaging technologies, and treatment innovations, ophthalmologists are more capable than ever in being able to accurately identify and effectively manage the rarest ocular diseases.
Still, attaining advice from seasoned clinicians can help newer ophthalmologists avoid the frustrations and complications that can accompany these uncommon conditions.
